Legacy route: /faqs/flagellum.html
Evolution of the bacterial flagellum
Evolution in (Brownian) space: a model for the origin of the bacterial flagellum
Copyright 2003 by N. J. Matzke
Version 1.0 (last updated November 10, 2003)
(Update section added September 2006.)
E-mail address: matzke@ATncseweb. (please remove obvious anti-spam modification)
|
Abstract: The bacterial flagellum is a complex molecular system with multiple components required for functional motility.� Such systems are sometimes proposed as puzzles for evolutionary theory on the assumption that selection would have no function to act on until all components are in place.� Previous work (Thornhill and Ussery, 2000, A classification of possible routes of Darwinian evolution. J Theor Biol. 203 (2), 111-116) has outlined the general pathways by which Darwinian mechanisms can produce multi-component systems. However, published attempts to explain flagellar origins suffer from vagueness and are inconsistent with recent discoveries and the constraints imposed by Brownian motion.� A new model is proposed based on two major arguments. First, analysis of dispersal at low Reynolds numbers indicates that even very crude motility can be beneficial for large bacteria.� Second, homologies between flagellar and nonflagellar proteins suggest ancestral systems with functions other than motility.� The model consists of six major stages: export apparatus, secretion system, adhesion system, pilus, undirected motility, and taxis-enabled motility.� The selectability of each stage is documented using analogies with present-day systems.� Conclusions include: (1) There is a strong possibility, previously unrecognized, of further homologies between the type III export apparatus and F1F0-ATP synthetase. (2) Much of the flagellum�s complexity evolved after crude motility was in place, via internal gene duplications and subfunctionalization.� (3) Only one major system-level change of function, and four minor shifts of function, need be invoked to explain the origin of the flagellum; this involves five subsystem-level cooption events.� (4) The transition between each stage is bridgeable by the evolution of a single new binding site, coupling two pre-existing subsystems, followed by coevolutionary optimization of components.� Therefore, like the eye contemplated by Darwin, careful analysis shows that there are no major obstacles to gradual evolution of the flagellum.� Contents:
Update, September 2006This essay has now been cited in the literature (Pallen et al. 2006, �Evolutionary links between FliH/YscL-like proteins from bacterial type III secretion systems and second-stalk components of the FoF1 and vacuolar ATPases.� Protein Science, 15(4), 935-941 - DOI) and linked from a peer-reviewed article I have just coauthored (Pallen and Matzke 2006, �From The Origin of Species to the origin of bacterial flagella.� Nature Reviews Microbiology, 4(10), 784-790. Advanced Online Publication on September 5, 2006 - DOI). Therefore, in order to avoid confusion, I will not update the text of this article at this address. I have, however, made some minor formatting changes, and updated the Reader Background page. While �Evolution in (Brownian) Space� was admittedly a first attempt, and I was a dedicated enthusiast rather than a professional, I think the model has stood up rather well over the last two and a half years. Writing in 2006, I would still agree with about 90% of the 2003 model. To summarize the major updates I would make: First, the hypothesis of homology between the Type 3 Secretion System export apparatus and the F1F0-ATPase (and its archaeal and eukaryotic equivalents) has been dramatically strengthened by the findings of two papers, Lane et al. 2006 (�Molecular basis of the interaction between the flagellar export proteins FliI and FliH from Helicobacter pylori.� Journal of Biological Chemistry, 281(1), 508-17 - DOI), and the aforementioned Pallen et al. 2006. As I predicted in 2003, sequence studies have now confirmed homology between FliH/YscL and F0-b (and its equivalents in other ATPases). They also strongly indicate that F1-delta is homologous to the C-terminal domain of FliH; I did not predict this, but it does further confirm my more general prediction of �a strong possibility, previously unrecognized, of further homologies between the type III export apparatus and F1F0-ATP synthetase.� However, I would retract some of my more speculative suggestions for ATPase homology to FliJ, FliO, and FliP (FliJ and FliO are apparently not even universally required in flagella). I am still hopeful regarding the suggestions for FliQ and FliR. Secondly, in the 2003 essay I for the most part assumed that the nonflagellar Type 3 Secretion System (NF-T3SS) was derived from the flagellum, rather than being an outgroup with a sister group relationship. I took this position partially to show that even under this assumption the evidence for evolution was strong, and partially because the evidence seemed to lean slightly in that direction. The parsimony argument of Pallen et al. 2005 and various minor points now have me leaning somewhat towards the view that the flagellar and nonflagellar systems are sister groups, and the NF-T3SS is therefore an outgroup. However, as we note in Pallen and Matzke 2006 the scientific community is split on this question. There are several avenues of investigation that might clarify matters, which I will explore in the future. Thirdly, the question of which proteins are actually universally �essential� for flagellum function, and which proteins have homology to other flagellar proteins or nonflagellar proteins, has been systematically reviewed in Table 1 of Pallen and Matzke (2006). I have reposted the table in my blog post on Panda's Thumb. It is important to note that this table is much more conservative than the Matzke 2003 homology suggestions, which ranged from well-established to loose speculation. The homologies in the 2006 table are all well-confirmed by standard BLAST techniques, except for five proteins where homology is based on structural or other similarities. Even for these five, two of the flagllar proteins have other known homologies based on sequence (FliC to FlgL and FliH to YscL), two are not universally essential (FliH and FliJ), and three of the homologies have been repeatedly put forward in the literature (FliC to EspA, FliK to YscP, and FliH/YscL to F0-b+F1-delta and equivalents). In the entire list, only one required protein has a new proposed homology that could be considered speculative (FliG, to MgtE). Many of the homologous and/or inessential proteins found in Table 1 of Pallen and Matzke 2006 were cited in the 2003 paper, but the 2006 table is an authoritative update and supercedes what is said here. The important overall point, as discussed in my blog post, is that of the 42 proteins in Table 1 of Pallen and Matzke, only two proteins, FliE and FlgD, are both essential and have no identified homologous proteins. This is substantially more impressive than the situation in 2003, and means that the evidence for the evolutionary origin of the flagellum by standard gene duplication and cooption processes is even stronger than in 2003. Important specific updates include: a homolog of FlgA has been confirmed (along the lines that I suggested in 2003); FliG has no homolog in NF-T3SS or the Exb/Tol systems, rather it may be homologous to the magnesium transporter MgtE; and the flagellar filament protein FliC (and its sister FlgL) is probably homologous to EspA and other pilus proteins found in NF-T3SS. I still suspect that all of the axial proteins (including FliE and FlgD) are homologous to each other and therefore to pilus proteins in NF-T3SS, but only the confirmed homologies are reported in Pallen and Matzke 2006. Finally, if I were doing a revision, I would update the terminology along the lines suggested in Desvaux et al. 2006 (�Type III secretion: what's in a name?� Trends in Microbiology 14(4), 157-160, April 2006 - DOI). As they point out, the terminological distinction between "flagellum" and "type 3 secretion system" is dubious and artificial, and it is more true to acknowledge that flagella have a type III secretion system. Therefore, there are two known groups of type III secretion systems, flagellar and nonflagellar, abbreviated F-T3SS and NF-T3SS. There is much more to be said about recent research and its implications for flagellum evolution. For the near future I intend to post my thoughts on this in the new flagellum evolution section of the Panda's Thumb blog. 1. Introduction1.1. A complex contrivanceThe bacterial flagellum is one of the most striking organelles found in biology.� In Escherichia coli the flagellum is about 10 μm long, but the helical filament is only 20 nm wide and the basal body about 45 nm wide.� The flagellum is made up of approximately 20 major protein parts with another 20-30 proteins with roles in construction and taxis (Berg, 2003; Macnab, 2003).� Many but not all of these proteins are required for assembly and function, with modest variation between species.� Over several decades, thousands of papers have gradually elucidated the structure, construction, and detailed workings of the flagellum.� The conclusions have often been surprising.� Berg and Anderson (1973) made the first convincing case that the flagellar filament was powered by a rotary motor. This hypothesis was dramatically confirmed when flagellar filaments were attached to coverslips and the rotation of cells was directly observed (Silverman and Simon, 1974).� The energy source for the motor is proton motive force rather than ATP (Manson et al., 1977).�� The flagellar filament is assembled from the inside out, with flagellin monomers added at the distal tip after export through a hollow channel inside the flagellar filament (Emerson et al., 1970).� The flagella of E. coli rotate bidirectionally at about 100 Hz, propelling the rod-shaped cell (dimensions 1x2 μm) 10-30 μm/sec.� The flagella of other species, powered by sodium ions rather than hydrogen ions, can rotate at over 1500 Hz and move cells at speeds of several hundred μm/sec.� The efficiency of energy conversion from ion gradient to rotation may approach 100% (DeRosier, 1998).� The bacterial flagellum is now one of the best understood molecular complexes, although numerous detailed questions remain concerning the function of various protein components and the exact mechanism of torque generation.� However, the origins of this remarkable system have hardly been examined.� This article will propose a detailed model for the evolutionary origin of the bacterial flagellum, along with an assessment of the available evidence and proposal of further tests.� That the time is ripe for a serious consideration of this question is discussed below. 1.2. An evolutionary puzzleBiologists find it almost inescapable to compare the bacterial flagellum to human designs: DeRosier remarks, �More so than other structures, the bacterial flagellum resembles a human machine� (DeRosier, 1998).� The impression is heightened by electron micrograph images (Figure 1) reminiscent of a engine turbine (e.g., Whitesides, 2001), and the scientific literature on the flagellum is filled with analogies to human-designed motors.� There is no shortage of authorities willing to express mystification on the question of the evolutionary origin of flagella.� In a 1978 review, Macnab concluded,
The basic puzzle is that the flagellum is made up of dozens of protein components, and deletion experiments show that the flagellum will not assemble and/or function if any one of these components is removed (with some exceptions).� How, then, could this system emerge in a gradual evolutionary fashion, if function is only achieved when all of the required parts are available?�
1.3. Theory: the evolution of systems with multiple required componentsThe standard answer to this question was put forward by Darwin.� Mivart (1871) argued that the �incipient stages of useful structures� could not have evolved gradually by variation and natural selection, because the intermediate stages of complex systems would have been nonfunctional.� Darwin replied in the 6th edition of Origin of Species (Darwin, 1872) by emphasizing the importance of change of function in evolution.� Although Darwin�s most famous discussion of the evolution of a complex system, the eye, was an example of massive improvement of function from a rudimentary ancestor (Salvini-Plawen and Mayr, 1977; Nilsson and Pelger, 1994), Darwin gave equal weight to examples of functional shift in evolution.� These included the complex reproductive devices of orchids and barnacles, groups with which he was particularly familiar (Darwin, 1851, 1854, 1862).� Intricate multi-component systems such as these could not have originated by gradual improvement of a single function, but if systems and components underwent functional shift, then selection could have preserved intermediates for a function different from the final one. The equal importance of improvement of function and change of function for understanding the evolutionary origin of novel complex systems has been similarly emphasized by later workers (Maynard Smith, 1975; Mayr, 1976).� Recent studies give cooption of structures a key role in the origin of feathers (Prum and Brush, 2002), and novel organs (Pellmyr and Krenn, 2002); Mayr (1976) gives many other examples.� Computer simulations also show the importance of cooption for the origin of complex systems with multiple required parts (Lenski et al., 2003). Do these common insights from classical, organismal evolutionary biology help us to understand the solution to the puzzle Macnab put forward regarding the origin of flagellum? Cooption at the molecular level is in fact as well-documented at it is at the macroscopic level (Ganfornina and Sanchez, 1999; Thornhill and Ussery, 2000; True and Carroll, 2002).� It has been implicated in origin of ancient multi-component molecular systems such as the Krebs cycle (Melendez-Hevia et al., 1996) as well as the rapid origin of multi-component catabolic pathways for abiotic toxins that humans have recently introduced into the environment, such as pentachlorophenol (Anandarajah et al., 2000; Copley, 2000), atrazine (de Souza et al., 1998; Sadowsky et al., 1998; Seffernick and Wackett, 2001), and 2,4-dinitrotoluene (Johnson et al., 2002); many other cases of catabolic pathway evolution exist (Mortlock, 1992).� All of these systems absolutely require multiple protein species for proper function.� Even for some molecular systems equaling the flagellum in complexity, reasonably detailed reconstructions of evolutionary origins exist. Generally these are available for systems which originated relatively recently in geological history, which are well-studied due to medical importance, and where phylogeny is relatively well resolved; examples include the vertebrate blood-clotting cascade (Doolittle and Feng, 1987; Hanumanthaiah et al., 2002; Jiang and Doolittle, 2003) and the vertebrate immune system (Muller et al., 1999; Pasquier and Litman, 2000). Thornhill and Ussery (2000) summarized the general pathways by which systems with multiple required components may evolve. They delineate three gradual routes to such systems: parallel direct evolution (coevolution of components), elimination of functional redundancy (�scaffolding,� the loss of once necessary but now unnecessary components) and adoption from a different function (�cooption,� functional shift of components); a fourth route, serial direct evolution (change along a single axis), could not produce multiple-components-required systems.� However, Thornhill and Ussery�s analysis did not distinguish between the various levels of biological organization at which these pathways might operate.� The above-cited literature on the evolution of complex molecular systems indicates that complex systems usually originate by a key shift in function of an ancestral system, followed by an intensive period of improvement of the originally crudely functioning design. At the level of the system, cooption is usually the key event in the origin of the modern system with the function of interest.� However, a great deal of the complexity in terms of numbers of parts is added to the system after origination.� These accessory parts get added by duplication and cooption of novel genes (for reviews of gene duplication in evolution, see Long, 2001; Chothia et al., 2003; Hooper and Berg, 2003) and/or duplication and subfunctionalization (Force et al., 1999) of genes already involved in the crudely-functioning system.� Cooption of whole subsystems, linking them to the �core� system, may also occur.� Therefore, improvement of function at the system level might be implemented by cooption at the level of a protein or subsystem.� Change of function at the system level might occur without any lower level cooption of new components.� Thornhill and Ussery�s four routes can be reduced to the two major pathways proposed by Darwin: improvement of current function (optimization) and shift of function (cooption).� Cooption remains its own category, while the other three routes (serial direct evolution, parallel direct evolution, and elimination of functional redundancy) can be considered as three versions of functional improvement, with the lower-level components undergoing optimization, coevolutionary optimization, or loss, respectively.� This conceptual framework is basically equivalent to the patchwork model for the evolution of metabolic pathways (Melendez-Hevia et al., 1996; Copley, 2000), where components are recruited from diverse sources and functional improvement or functional shift might occur at any organizational level, e.g. system, subsystem, protein, or protein domain.� 1.4. Constructing and testing evolutionary modelsIn order to explain the origin of a specific system such as the flagellum, the general theory discussed above must be combined with the available evidence in order to produce a detailed, testable model.� Detail in evolutionary scenarios makes them more testable, not less: Cavalier-Smith argues that �Specifying transitional stages in considerable detail is not unwarranted speculation, but a way of making the ideas sufficiently explicit to be more easily tested and rigorously evaluated� (Cavalier-Smith, 2001b).� Obviously �detailed� cannot mean that every mutation and substitution event be recorded � for events that occurred billions of years ago this is impossible.� A detailed evolutionary model should reduce a puzzling event like the origin of the flagellum into a series of events that occur by well-understood processes.� In an ideal model, the origin of every protein component will fulfill three criteria.� First, a putative ancestral protein with a different function (a homolog that can reasonably be suspected to precede the flagellum) should be identified.� Second, the cooption of the protein should occur by a reasonably probable mutation event -- e.g., a mutation produces a single new binding site enabling one protein to act on another.� Initially this new complex functions crudely, but can gradually be perfected by coevolutionary optimization of the two proteins.� Third, the selective regime favoring retention of the coopted protein should be identified.� Each of these three criteria encourages further testing against new data. Hypothesized homologies can be assessed by new data, for example by detailed sequence analysis or the comparison of protein structures.� The plausibility of mutational steps can be investigated by examination of similar mutations observed today; and the selective forces invoked can be assessed by study of analogies and by mathematical modeling.� Furthermore, an evolutionary model might have testable implications for other fields: for example, if a biological system is hypothesized to be derived from a homologous system, similarities in mechanism between the two systems would be suspected.� The fact that we do not have all of the data that we would like, and that uncertainty is high, are not problems unique to evolutionary models; rather, these problems are commonplace in any advancing science.� For example, many contradictory models have been published for the mechanism of motor action in the flagellum, and most (or all) of them must be wrong, but this has not stopped anyone from proposing new models (Schmitt, 2003).� Science is advanced by proposing and testing hypotheses, not by declaring questions unsolvable. 2. Background2.1. Modern flagellaThe canonical flagellum of E. coli is shown in Figure 2.� Descriptions of the structural components are given in Table 1.� Cytoplasmic components involved in regulation and assembly, as well as the chemotaxis components, are listed in Table 2.� Excellent overviews of flagellar function and assembly are available elsewhere (Berg, 2003; Macnab, 2003) and so will not be discussed further here.
|

|
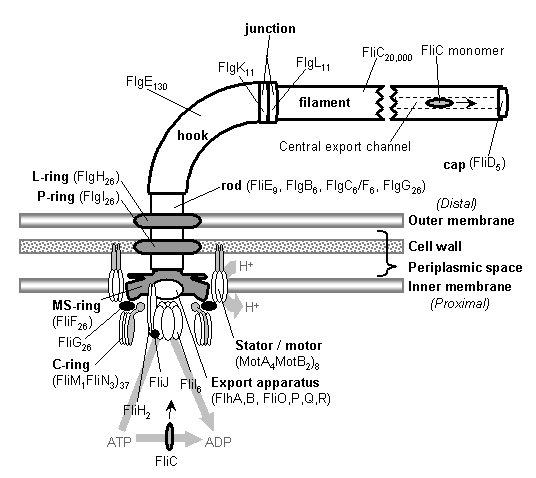
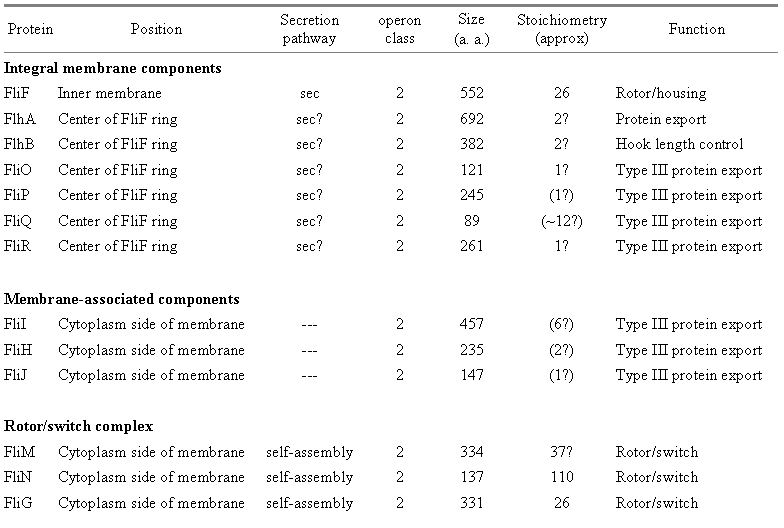
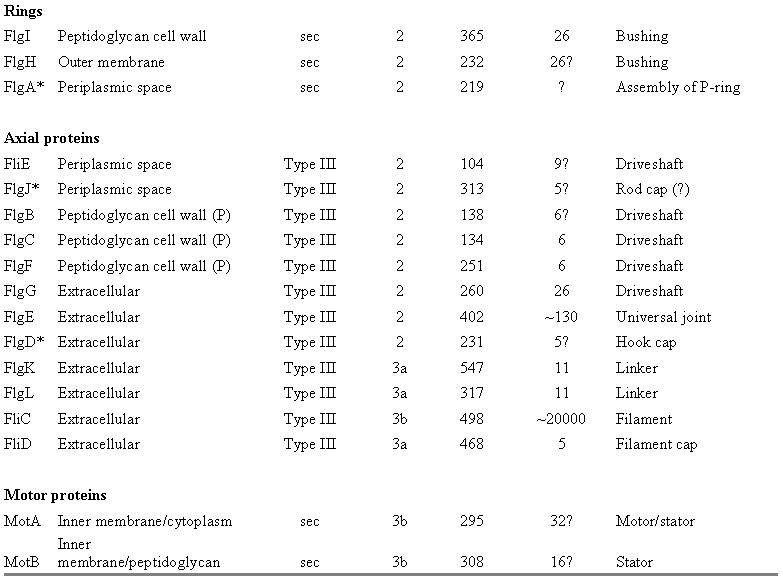
Table 1: Structural components of the E. coli flagellum.� Based on recent reviews (Berg, 2003; Macnab, 2003); figures in parentheses represent suggestions made in this paper.� Components with an asterisk (*) are not included in the final structure.
|
|
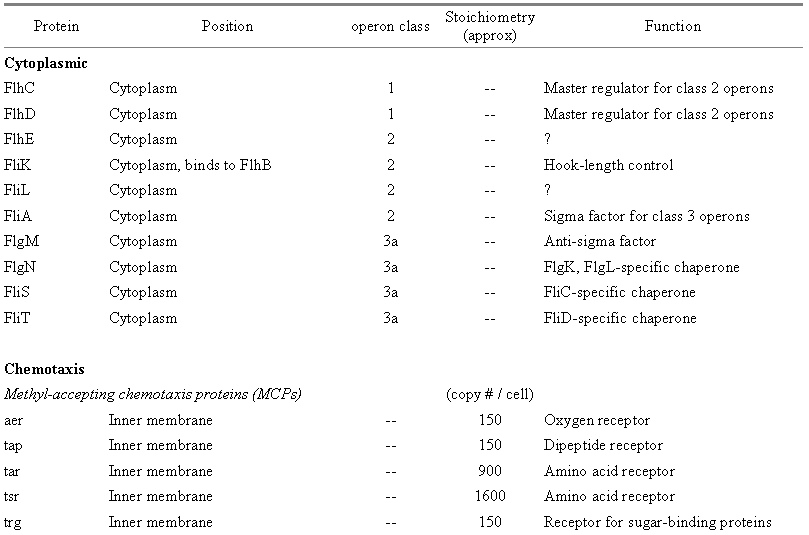
Table 2: Components of the E. coli regulation/assembly and chemotaxis systems.� Cytoplasmic components based on Berg (2003) and Macnab (2003), chemotaxis components based on Eisenbach (2000).
|

2.2. Previous attempts to explain flagellar origins
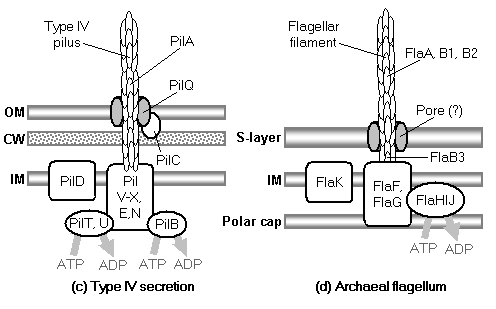
Occasional examples of very general suggestions about the evolutionary origin of flagella can be found in the literature, for example in discussions of how various aspects of the chemotaxis system are optimized (Berry, 2000); in the suggestion that prokaryote flagella may have been a relatively late invention, after biofilms and microbial mats had become well-developed and crowding on surface habitats became a problem (Stoodley et al., 2002); or in the alleged common ancestry of archaeal and bacterial flagella (Harshey and Toguchi, 1996).� Archaeal and bacterial flagella were indeed once thought to be homologous (Jones et al., 1987), but they are actually totally distinct motility systems (Jarrell et al., 1996; Faguy and Jarrell, 1999; Thomas et al., 2001).� Although both kinds of flagella rotate and are superficially similar, archaeal flagella are fundamentally different in many respects (Table 3).� In archaeal flagella, the filaments are thinner, lack a central channel, and subunits are added from the base rather than the tip. Forward movement is typically attained by clockwise rather than counterclockwise motion.� Additionally, archaeal flagella are probably powered by ATP rather than protonmotive force (suggested by homologies of FlaI to PilT/U (Jarrell et al., 1999; Thomas et al., 2001; Merz and Forest, 2002, although the literature is contradictory: Bardy et al. (2003) assert that archaeal flagella use protonmotive force, but cite no supporting evidence). Finally, the homologies of the two flagella to nonflagellar secretion systems are different.� The bacterial and archaeal flagella are therefore a classic case of analogy, not homology (Faguy et al., 1994; Jarrell et al., 1996; Bayley and Jarrell, 1998; Faguy and Jarrell, 1999; Thomas et al., 2001; Thomas et al., 2002; Bardy et al., 2003).� However, the misperception persists in the assumption that the flagella (Harshey and Toguchi, 1996; Campos-Garcia et al., 2000; Rizzotti, 2000) or their basal bodies (Cavalier-Smith, 2002a, 2002c) are homologous. On the other hand, the chemotaxis systems are indeed homologous, and are shared with nonflagellar motility systems as well (Faguy and Jarrell, 1999; Koretke et al., 2000).�
|
Table 3: Some microbial motility systems. Several more mysterious systems (the perhaps cytoskeleton-based motilities of Mycoplasma and Spiroplasma; Trachtenberg et al., 2003) have been excluded. Prokaryotes undoubtedly have additional motility systems that have not yet been discovered.� Only one eukaryote system, the cilium or eukaryotic flagellum, is included in the table, because it is often confused with the prokaryote systems even though it is totally distinct.� Many other eukaryote motility systems, not relevant here, are not listed. Data gathered from many sources (Young et al., 1999; Eisenbach, 2000; McBride, 2001; Thomas et al., 2001; Bardy et al., 2003; Youderian et al., 2003).
|

A slightly more detailed attempt at explaining the origin of the bacterial flagellum was made by de Duve (1995), who apparently got the bacterial flagellum confused with the completely different eukaryotic cilium (also known as the eukaryotic flagellum or undulipodium in an interminable terminological dispute; see Corliss, 1980; Margulis, 1980; Cavalier-Smith, 1982).� He suggested that the flagellum, which he acknowledges is rotary, was somehow descended from a simpler ATP-powered filament-bending motor. In a more reasonable vein, de Duve then gave a brief scenario for the gradual origin of chemotactic behavior from random swimming, but was again puzzling in postulating that essentially fully functional, bidirectional-switching flagella with specific positioning on the cell surface existed before the signal transduction system was coupled to the flagellum.� What the purpose of switching would be without a chemotaxis system was not explained. De Duve furthermore stated that these well-developed but non-chemotactic flagella gave �little advantage� until they were chemotactically enabled, leaving unexplained the selective reason for the origin of the whole nearly-complete system in the first place.�
Finally, Goodenough (1998; 2002) offers a short account deriving a flagellum from a proton-transducing membrane channel. She postulates that a coopted protein increased the efficiency of proton transport, and rotated the channel as a by-product.� Later binding of a filament to the outside of this rotating channel produced primitive motility which increased food gathering ability.� However, the original function of proton transport (which, uncoupled to another process, would simply de-energize the cytoplasmic membrane) is not specified. In her 2002 account Goodenough suggested that a fibrous protein binding to the F1F0-ATP synthetase produced the proto-flagellum.� Presumably she meant that the proto-filament would bind to the distal side of a c-subunit of F0.� As recent work indicates that F0-c and F1-εγ rotate inside the F0-ab and F1-αβδ complex (Weber and Senior, 2003), Goodenough�s suggestion is not immediately impossible, but suffers difficulties similar to those discussed for Rizzotti (2000), below.
Cavalier-Smith is one of the few who has proposed detailed hypotheses for� the origin of many fundamental features of eukaryotes and prokaryotes (Cavalier-Smith, 1987a, 1987b, 2001a, 2002b, 2002a, 2002c).� He bases his work on a refreshingly clearly-stated philosophy for reconstructing the origin of complex systems, advocating a holistic approach considering environment, organism, mutation, and selection all together and emphasizing testability (Cavalier-Smith, 2001a).� Although Cavalier-Smith has addressed the origin of the eukaryotic cilium on several occasions (Cavalier-Smith, 1978, 1982, 1987b, 2002b), Cavalier-Smith�s only treatment of the origin of the bacterial flagellum is found in a 1987 article (Cavalier-Smith, 1987a).� He makes two suggestions: first, that a mutant version of an outer membrane protein pore formed a tubular polymer extending through the outer membrane into the extracellular medium.� Linking this to proton-conducting proteins in the cytoplasmic membrane provided the primitive motor.� In this scheme, spirochete axial filaments were derived from regular flagella. His second suggestion was that flagella evolved from gliding motility systems, which are also widespread and powered by protonmotive force.� Some early models of gliding motility postulated a spirochete-like mechanism, with rotating filaments in the periplasmic space, and on this basis spirochetes might represent a transitional stage.� Motility would develop from rotating filaments first used just to stir the fluid in the periplasmic space and increase diffusion of nutrients.� On either scenario, the rotary mechanism existed from the beginning of the evolutionary sequence, and the first crude motility function would have been selected for because it increased random dispersal, useful in overcrowded regions depleted in nutrients.� Much of the complexity could have post-dated the original crudely functioning motility.
Cavalier-Smith was hampered by the relatively primitive state of knowledge at the time, and he conceded that the actual evolutionary process must have been much more complicated than his suggestions.� The linkage between the filament and motor is very complex, mediated by about ten proteins, and the filament subunits are secreted through the base of the flagellum via a type III export pathway, rather than via a type II pathway as might be expected for a protein derived from an outer membrane pore; type III virulence systems do utilize an outer membrane secretin secreted by the type II pathway, and the flagella P- and L-ring proteins FlgI and FlgH are similarly secreted via the type II pathway (Macnab, 2003).� A secretin might therefore be more likely posited as the source for FlgH; this will be discussed in more detail below.
Regarding the postulated homology between gliding motility and the axial filaments of spirochetes, today it is apparent that gliding motility is not a matter of rotating periplasmic filaments.� Two mechanisms for gliding motility have been clearly identified (Merz and Forest, 2002; Bardy et al., 2003).� First, the social gliding of Myxococcus xanthus occurs via retraction of type IV pili, sometimes also called twitching motility (Merz and Forest, 2002).� Second, the adventurous motility of M. xanthus is driven by the secretion of a polysaccharide gel (slime) via the junctional pore complex; a similar complex is found in gliding cyanobacteria.� The mechanism of the gliding motility of Cytophaga and Flavobacterium is still a matter of speculation (McBride, 2001), but may involve a ratchet structure and slime secretion (Bardy et al., 2003).� These latter forms of gliding motility inspired the comparison between flagella and gliding motility as they are powered by protonmotive force, and beads attached to the cell surface of Cytophaga will rotate (Eisenbach, 2000).� Thus, it is occasionally suggested (Cavalier-Smith, 2002a), even in textbooks (e.g. Campbell, 1993), that flagella and gliding motility are homologous, and the gliding motility apparatus may be some version of the flagellum basal body without the flagellar filament.� As our understanding of slime-related gliding motility is still limited (the relevant genes are still being identified, much less detailed mechanism or structure), the possibility of any connection between type III protein secretion and polysaccharide secretion is difficult to evaluate.� However, the study of gliding motility bears close watching: the recent discovery of homology between M. xanthus gliding motility proteins AglS/AglV to TolR and of AglR/AglX to TolQ (Youderian et al., 2003) which are in turn homologs of the flagellar motor proteins MotA and MotB (Cascales et al., 2001) suggests that there may be a common mechanism for coupling proton flow to motility.� If the general similarity between the junctional pore complex and type III secretion systems (Spormann, 1999; Merz and Forest, 2002) turns out to be more than skin deep, then the common descent of gliding motility and flagella from an ancestral motility organelle will have to be seriously considered. Cavalier-Smith�s suggestion that stirring the periplasmic fluid may have been a precursor to primitive motility is similar to Rizzotti�s main suggestion and will be discussed in the next section.
The only major recent attempt at explaining the origin of the flagellum is that of Rizzotti (2000), which, like Goodenough, proposes that the flagellum was derived from the F1F0 ATP synthetase.� The initial appeal of this hypothesis derives from the spate of recent comparisons between the flagellum and ATP synthetase as proton-driven, rotary motors (Block, 1997; Boyer, 1997; Khan, 1997; Sabbert and Junge, 1997; Berg, 1998; Oplatka, 1998a, 1998b; Berry, 2000; Walz and Caplan, 2002), sometimes leading to the suggestion of homology (Oster and Wang, 2003).� These comparisons go back at least to Cox et al.�s (1984) proposal that the ATP synthetase had a rotary mechanism, and continued through the testing and refinement of this hypothesis (Mitchell, 1985; Sabbert and Junge, 1997; Weber and Senior, 2003), followed by the conclusive demonstration of rotation by direct observation of an actin filament tethered to the gamma subunit of F1-ATPase (Noji et al., 1997).� A relationship between the F1F0 ATP synthetase and the flagellum is further suggested by homology between the flagellar ATPase FliI and the β subunit of F1-ATPase, indicated by ~30% sequence similarity (Albertini et al., 1991; Vogler et al., 1991).� The α and β subunit ATP synthetase subunits are themselves paralogous, with only the β subunit retaining catalytic activity (Gogarten et al., 1989; Gogarten and Kibak, 1992).
In a creative scenario (Figure 3),� Rizzotti imagined that an accidental insertion in the middle of the F1-γ subunit created a short filament outside the cytoplasmic membrane, between the membrane and the cell wall.� As the synthetase subunits rotated, this protofilament served to mix the nearby fluid, increasing the diffusion of molecules in and out of the cell.� This provided sufficient selective benefit to retain the mutation.� Production of a more sophisticated mixing instrument occurred via duplication and modification of the mutant γ subunit, so that branches of the filament extended above the cell wall.� In the process, the ε and δ subunits were lost, along with ATPase activity, resulting in a proton-powered stirring mechanism with incipient motility function.� From here, a process of optimization ensued.� Selection first favored random motion of the cell that further improved nearby fluid mixing and diffusion.� More powerful motility followed by extension of the filament and by duplications of the proton-transmitting proteins of the stator (in this scenario, derived from the c subunit of the F0 structure).� The F1-αβ complex apparently became the rotor inside the stator ring.� Rizzotti concluded by discussing a number of other steps that must have happened along the way, although the order is not specified.� However, it seems that he considered the origin of the export apparatus a relatively late event.� Rizzotti hypothesized that once the central cavity became large enough, a secretion complex (presumably a type III export apparatus already functioning elsewhere) was patched in at the base of the rotor, allowing the secretion of a more complex filament.
|
|
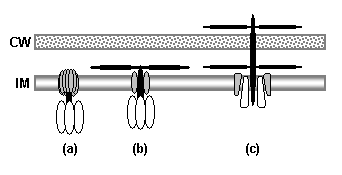
Rizzotti argued that bacteria with a single membrane were simpler and therefore probably ancestral to gram-negative bacteria with both an inner and outer membrane.� He hypothesized that the outer membrane arose as an alimentary adaptation from extensions of the inner membrane.� The L- and P-rings arose as the developing outer membrane encroached on the flagellum (gram positive bacteria, lacking outer membranes, have no requirement for the L- and P-rings and lack them altogether).� Rizzotti discounted the alternative scenario, whereby the flagellum arose in a bacterium already possessing a double membrane, because he deemed the simultaneous origin of the rings and filament too difficult.�
This scenario is considerably more detailed than any other available, but remains vague on the specific origin of almost all of the proteins that make up the flagellum.� Although Rizzotti does make use of some interesting similarities between the flagellum and ATP synthetase, and he is able to come up with a proposal that includes rotary motion from the beginning, there are major flaws which shall be discussed shortly.� Before the critique, however, it is worth noting that Rizzotti�s scenario has been cited by Cavalier-Smith (2001a) as well as others (Rosenhouse, 2002), apparently for lack of anything better.
Rizzotti�s suggestion that stirring might be a primitive function of a proto-flagellum is intuitively appealing, but intuition is a poor guide to life at a low Reynolds number (Purcell, 1977; Vogel, 1994; Purcell, 1997).� Bacteria live in a world dominated by Brownian motion, where viscous forces overwhelm inertia and small molecules spread much faster by diffusion than by bulk movement of fluid.� The scale at which moving fluid (stirring) or moving through fluid (swimming) will increase diffusion into the cell is determined by comparing the time for transport by diffusion (td) versus the time for transport by bulk flow such as stirring (ts) (Purcell, 1977).� For diffusion, the average time td for transport of a particle a distance l, with diffusion coefficient D is (Berg, 1993):
|
| (1) |
while the corresponding time for bulk flow transport via stirring (ts) is approximately (Purcell, 1977):
|
| (2) |
that is, the distance l divided by the fluid velocity v induced by stirring.� Stirring �works� only if the transport time using stirring is less than the transport time from simple diffusion:
|
| (3) |
|
| (4) |
|
| (5) |
The ratio in equation (5) gives the P�clet number, P�, which must be greater than unity for bulk flow to have substantial impact on diffusion (Vogel, 1994).� For a typical small molecule (e.g. sucrose) in water, D=10-10 m2s-1.� For a typical-length bacterium (1 μm) moving fluid past itself with the swimming velocity of a typical fully functional flagellum (30 μm/s), P� = 0.06 << 1 (Vogel, 1994).� For Rizzotti�s primitive stirrer, P� would be even lower.� As Purcell (1977) noted, in the world of low Reynolds number, �stirring isn�t any good�. Bacteria that do induce currents for their benefit (e.g., Thar and Kuhl, 2002) probably succeed because of the large number of bacteria cooperating in the effort, in effect increasing body size.� Another postulated function of primitive motility, swimming for the sake of running into more molecules, also does not work: Purcell calculated that a bacterium would have to swim 700 μm/sec in order to gather only 10% more food molecules. Thus, if diffusion of molecules into the cell is the only matter of concern, a bacterium will do just as well by sitting still as it will by stirring or swimming.� The reason bacteria swim is not to increase diffusion but to find locations with a higher local concentration of nutrient molecules (Purcell, 1977; Berg, 1993; Vogel, 1994).� Purcell�s argument breaks down in situations where the uptake rate parameter, a, representing the fraction of available molecules being consumed each second, is greater than 1 s-1.� However, a typical value for a is 0.01, where uptake is considered negligible (Dillon et al., 1995; Mitchell, 2002).� Thus, fundamental physical considerations make the hypothesized stirring filament an unlikely intermediate.
Additional difficulties with Rizzotti�s model exist.� While it is unrealistic to expect sequence similarity to give evidence for the ancestry of every component of the 3+ billion year old flagellum, considering the time lapse and large nature of some of the changes that must be postulated on any scenario, a scenario certainly should not contradict those homologies that have been identified.� The Rizzotti scenario (Figure 3) implies homology between the synthetase F1-αβ subunits and FliF/FliG (the flagellar rotor), but the homology that inspired the scenario is between F1-αβ and FliI (the ATPase that energizes export of rod, hook, and filament). Similarly, Rizzotti (2000) implies that the F0-c subunit is homologous with the flagellar motor proteins MotAB, but sequence homology has instead been discovered homology between MotAB and a phylogenetically widespread family of proteins that couple protonmotive force to diverse membrane transport processes.� These homologs, namely ExbBD (Kojima and Blair, 2001) and TolQR (Cascales et al., 2001), provide a simpler and much more direct ancestor for MotAB.� The homologies could be explained by invoking additional independent cooption events, but this would require a rather more complex scenario than that presented by Rizzotti.
As Rizzotti�s scenario fails on the twin tests of homology and a simple model of stirring at a low Reynolds number, it is now time to see if Rizzotti can be improved upon.� It should be noted that although published proposals about flagellar evolution are very limited, the topic is a popular one as the flagellum is the icon of the antievolutionary �Intelligent Design� movement.� Therefore several of the ideas proposed here have been previously raised in informal debates about flagellar evolution.� Miller (2003, 2004) and Musgrave (2004) review this aspect of the debate in detail, and Musgrave proposes a model that is similar in outline to that presented here, although his account is more general.
3. The Model
3.1. Phylogenetic context and assumed starting organism
The paradigm for prokaryote phylogeny, if there is one, is the universal rRNA tree.� This shows a number of widely separated bacterial lineages, with archaea and eukaryotes separated from them all by a very long branch. This tree is unrooted, and many possible rootings have been proposed in the literature.� As these are the most remote and difficult phylogenetic events it is possible to study, and as there is by definition no outgroup to life in general, the debate can be expected to continue for some time.� For current purposes the most important point is that flagella are widespread across the bacterial phylogenetic tree, with losses in various taxa and no clearly primitive nonflagellate taxa.� It is therefore assumed that flagella evolved near the base of the bacterial tree.�
Rizzotti (2000) and others (e.g., Koch, 2003) have suggested that the last common ancestor of bacteria was gram positive.� However, the very general consideration that most of the bacterial phyla are gram negative, including the many different taxa that come out as basal on different analyses, weighs against this hypothesis. Therefore, we shall side with Cavalier-Smith, who argues that the last common ancestor was gram-negative.� He has put forward the most detailed model for the origin of bacteria and the double membrane (Cavalier-Smith, 2001a, 2002a).� The model thus begins with a generic double-membraned, gram-negative bacterium.� Whether or not archaea are an outgroup to extant bacteria (the most common opinion), or a relatively late group derived from actinobacteria (high G+C content gram-positive bacteria), in turn derived from endobacteria (low G+C-content gram-positives) and cyanobacteria (Cavalier-Smith, 2002a) shall be left unresolved, although implications of flagellar evolution for Cavalier-Smith�s scheme will be highlighted.� The present model will begin with a reasonably complex bacterium, already possessing the general secretory pathway and type II secretion system, as well as signal transduction, a peptidoglycan cell wall, and F1F0-ATP synthetase.� As these components are ubiquitous, almost certainly predating the cenancestor, whereas many bacteria (perhaps 50% of species) lack flagella entirely, this seems plausible.� These assumptions are consistent with Cavalier-Smith�s position that the cenancestor was a bacterium similar in complexity to modern bacteria (Cavalier-Smith, 2001a, 2002a). Cavalier-Smith (2002a) hypothesizes that chlorobacteria may be the most basal offshoot of the tree and be primitively nonflagellate.�
3.2. Starting point: protein export system
3.2.1. Type III secretion systems
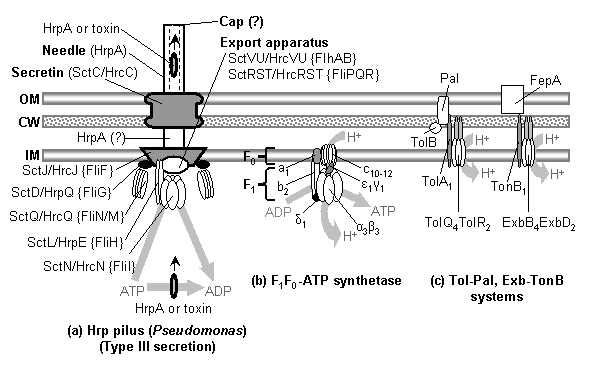
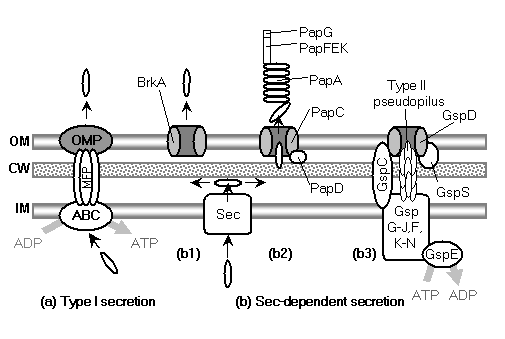
The model begins with a hypothetical primitive type III export apparatus.� As terminology is sometimes inconsistently used, following Hueck (1998), the term �secretion� is reserved for the transport of proteins from the cytoplasm to the cell surface or the extracellular medium.� �Export� refers to the transport of proteins from the cytoplasm to the periplasmic space.� An export system plus a mechanism to cross the outer membrane forms a secretion system. Bacteria make use of a number of distinct secretion systems, reviewed as a group elsewhere (Hueck, 1998; Thanassi and Hultgren, 2000a; van Wely et al., 2001).� Six major well-characterized secretion systems (Figure 4a, Figure 5) are reviewed by Thanassi and Hultgren (2000a).� These are: (1) autotransporters (Henderson et al., 1998), (2) the chaperone/usher pathway (Thanassi et al., 1998), (3) type I secretion or the ATP-binding cassette (ABC) transporter (Buchanan, 2001), (4) type II secretion or general secretory pathway (Pugsley, 1993; Sandkvist, 2001; Cao and Saier, 2003), (5) type III secretion systems of flagellar export and some infectious systems (Hueck, 1998; Cornelis and Van Gijsegem, 2000), and (6) type IV secretion (Christie and Vogel, 2000; Christie, 2001), homologous to type II secretion, conjugation pili, twitching motility systems, and archaeal flagella (Jarrell et al., 1996; Bayley and Jarrell, 1998; Sandkvist, 2001; Peabody et al., 2003).� It is likely that systems will be added to the list in time.�
|
|
|
|
About 10 well-conserved protein species make up the core of the type III export apparatus, which is used to export the axial components of bacterial flagella (rod, hook, filament, adaptor, and cap proteins).� In 1994 it was discovered that homologs of these proteins are also used to secret virulence factors in a diverse array of proteobacterial pathogens, such as Yersinia pestis, Salmonella typhimurium, Pseudomonas aeruginosa and enteropathogenic E. coli (Hueck, 1998).� The term �type III secretion system� is commonly used to refer to the virulence systems, but here it will be used to denote the class of secretion systems that make use of the type III export pathway.� This includes the two currently known members (virulence and flagellar secretion systems) and any unknown homologs.
The existence of a nonflagellar type III export apparatus falsifies the argument that flagellar components are useless if they are not part of a fully functioning flagellum. One answer to Macnab�s (1978) query, �What advantage could derive�from a �preflagellum� (meaning a subset of its components)� is now obvious: a subset of flagellar components could serve as an export system.� Thus, the model for the origin of flagella begins with the hypothesis of a primitive type III export system.� This hypothesis, however, requires justification on several grounds in order to ameliorate obvious objections.
3.2.2. Are nonflagellar type III secretion systems derived from flagella?
The fact that known nonflagellar type III secretion systems are restricted to proteobacteria, and that these systems are mostly virulence systems specializing on eukaryotes (which are probably far younger than flagella), lead Macnab (1999) as well as others (He, 1998; Kim, 2001; Plano et al., 2001) to conclude that the flagellar pathway is probably the older one, and that type III virulence systems are derived from flagella.� Although some apparently avirulent type III secretion systems have been discovered (e.g., in the legume symbiote Rhizobium; see Marie et al., 2001), and the phylogenetic distribution of type III secretion systems has been widened somewhat by their discovery in Chlamydiales (Kim, 2001), these data still support the conclusion that type III virulence systems are derived eukaryote-interaction systems, rather than phylogenetically basal homologs.� Phylogenetic analysis of type III secretion systems seemed to confirm the case (Nguyen et al., 2000).� Aizawa (2001) was one of the few dissenting opinions, arguing that flagella and virulence systems might have diverged in parallel from a common nonflagellar ancestor, pointing out that there are bacteria that parasitize or prey on other bacteria, a point with some merit although predatory bacteria are poorly studied (Guerrero et al., 1987).
Nguyen et al.�s (2000) conclusion has recently been challenged by Gophna et al. (2003), who demonstrated with phylogenetic trees of FlhA, FliI, FliP, and FliO homologs that type III virulence system sequences do not nest within flagellar sequences.� This supports the view that the two systems diverged from a common ancestor, which could plausibly have been a type III export system functioning in a nonflagellar, nonpathogenic context. However, Gophna et al. (2003) are not able to exclude the possibility that virulence systems evolve more rapidly, or that the frequent lateral transfer of type III virulence system genes (Nguyen et al., 2000; Gophna et al., 2003) might have increased the rate of sequence divergence.� Gophna et al.�also cite for support the progressionist notion that evolution disfavors events such as the simplification of complex systems like the flagellum, a dubious proposition in modern evolutionary theory, especially considering the common evolutionary trend of simplification in pathogens and parasites.� As long as known nonflagellar type III secretion systems are phylogenetically restricted and only function as specialized systems for eukaryote penetration, the suspicion will remain that they are derived from flagella.� For the purposes of the current discussion it will be assumed that type III virulence systems are derived, although they still give valuable insights about the possible traits of a hypothetical ancestral type III secretion system.
3.2.3. An ancestral type III secretion system is plausible
If type III virulence systems are derived from flagella, what is the basis for hypothesizing a type III secretion system ancestral to flagella?� The question would be resolved if nonflagellar homologs of the type III export apparatus were to be discovered in other bacterial phyla, performing functions that would be useful in a pre-eukaryote world.� That such an observation has not yet been made is a valid point against the present model, but at the same time serves as a prediction: the model will be considerably strengthened if a such a homolog is discovered.� For the moment, it is easy enough to explain the lack of discovery of such a homolog on the basis of lack of data.� Knowledge of microbial diversity is quite poor (Whitman et al., 1998): far less than 1% of bacteria extant in a particular environment are readily culturable (Hayward, 2000). Cultivation-independent surveys of prokaryote diversity based on environmental rRNA sequencing commonly discover deeply-branching microbes previously unknown to science (DeLong and Pace, 2001), and that certain groups are unexpectedly ubiquitous (Karner et al., 2001).� In addition, only a fraction of cultured microbes have been studied in any substantial biochemical or genetic detail, and this subsample is heavily skewed towards pathogens and convenient model organisms. Of the ~112 complete bacterial genomes sequenced as of July 2003 (http://www.ncbi.nlm.nih.gov/PMGifs/Genomes/eub_g.html), at least two-thirds are pathogens, mutualists, or commensals of multicellular eukaryotes.� Many of the free-living bacteria that have been sequenced are extremophiles or are used in industrial applications.
Even with such a skewed dataset, a general argument for the plausibility of a primitive type III export system can be constructed on the basis of analogy.� Each of the six secretion systems described above has been coopted to serve diverse functions by prokaryotes (Table 4).� The thoroughness of some of the observed convergences is remarkable � notably, all of the systems have been adapted for eukaryotic virulence, five secrete surface structures, at least four are used for adhesion, three or four form pili, and two perform motility-related functions. That pili and adhesion often play a role in virulence in well-studied organisms is not particularly significant, as such functions are useful in free-living contexts as well (Kennedy, 1987).� The overall picture is that any secretion system that exists will sooner or later get coopted for diverse functions, including virulence, in various lineages.� The commonality of the virulence function in known systems almost certainly reflects human interests rather than the situation in the wild.
|
Table 4: Convergent functions of well-characterized prokaryote secretion systems.� Other secretion systems are known to exist: e.g., curli fimbriae based on the extracellular nucleation/precipitation pathway (Smyth et al., 1996; Wu and Fives-Taylor, 2001; Chapman et al., 2002) and slime secretion (Merz and Forest, 2002).� Others undoubtedly remain to be discovered.
|


It might be objected that with so many available secretion systems, postulating the existence of an additional system is superfluous.� However, many bacteria have multiple secretion systems.� An illustrative case is Pseudomonas aeruginosa, which has all of the above-listed systems (Bitter, 2003). Furthermore, many bacteria will have two or more copies of certain types of secretion systems, with mildly to strongly divergent functions: e.g., E. coli can have both P-pili and type 1 pili (Thanassi and Hultgren, 2000a); Salmonella and Yersinia have two type III virulence systems each (Cornelis and Van Gijsegem, 2000); and Pseudomonas aeruginosa has at least two type II secretion systems and probably two kinds of type IV pili (Bitter, 2003).
3.2.4. The origin of a primitive type III export system
Type III virulence systems have well-conserved homologs of the following flagellar components (Plano et al., 2001): FliF (the membrane-embedded MS-ring); FlhA, FlhB, FliP, FliQ, FliR (integral membrane export components inside the MS-ring); FliI and FliH (ATPase and regulator); and FliG and FliM/N (the switch complex).� The primitive type III secretion system would not necessarily have had all of the components that are conserved in the possibly derived virulence systems.� In particular, if the type III virulence systems are derived, the homologs of the switch complex proteins (FliN/M, FliG) are probably retained only in order to stabilize/support the coadapted secretion complex and FliF ring, and are otherwise vestigial.�
FliF is fundamentally a membrane pore and so its origin must lie with the origin of transport proteins in general, a question explored by Saier (2003).� FlhA and FlhB are larger than FliOPQR, and have large cytoplasmic C-terminal domains that appear to bind the export substrates. FlhA interacts with FliF and the soluble components of the type III secretion system but its exact function is unknown.� FlhB plays a key role in determining whether rod/hook or filament axial proteins are secreted, and therefore controls the length of the hook by a poorly-understood mechanism (Macnab, 2003).� Substrate switching would not have been a necessary feature of a primitive type III secretion system, but perhaps the association of proto-FlhA and/or FlhB with the proto-FliF pore turned it from a somewhat general passive transporter into a substrate-specific passive transporter. One of the differences between type II and type III secretion systems is that type II systems recognize their substrates by a N-terminal signal peptide that is removed during transport.� The signal sequences for type III secretion substrates are also in the N-terminal regions but they are not cleaved (B�ttner and Bonas, 2002).� Perhaps this difference allowed the primitive type III secretion system to export an important substrate on a different control circuit independent of the sec pathway, and this finer control was the selective basis for the retention of the system.
3.2.5. The relationship between type III export and the F1F0-ATP synthetase
That a phylogenetically basal type III export apparatus must have existed is supported by several additional facts.� As discussed previously, the protein that powers protein export in type III secretion, FliI, has long been considered homologous to the F1 subunit of F1F0-ATP synthetase on the basis of about 30% amino acid identity to the active F1-β subunit (Albertini et al., 1991; Vogler et al., 1991; Gogarten et al., 1992).� The F1-αβ ATPase is a heterohexamer made up of alternating α-subunits (noncatalytic) and β-subunits (catalytic).� This pattern is shared by all bacteria and is also found in the archaeal A-ATP synthase and eukaryote V-ATP synthase, so F1-α and F1-β are thought to have diverged before the cenancestor (Gogarten and Kibak, 1992).� FliI, on the other hand, probably consists of a homohexamer of catalytic subunits (FliI�s hexameric nature was only recognized very recently: Blocker et al., 2003; Claret et al., 2003).� It diverges before the F1-α and F1-β split in sequence similarity trees, and thus probably also diverged prior to the cenancestor (Gogarten and Kibak, 1992).� However, it is more similar to the F1 subunits than the more distantly related hexameric ATPases such as the RNA/DNA helicase termination factor rho (Boyer, 1997), and therefore Gogarten and Kibak (1992) conclude that the FliI family diverged specifically from a primitive F1-ATPase prior to the cenancestor.� There is not similar evidence that flagella specifically evolved before the cenancestor, so this is a point in favor of the primitive type III export system hypothesis.
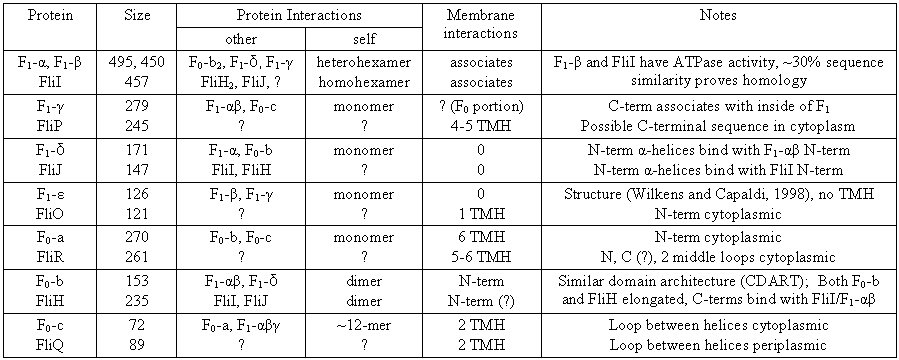
In light of the long-established homology between FliI and F1-αβ, it is surprising that there have been few searches for further homologies between the F1F0-ATP synthetase and type III export system.� Sequence similarity searches do not turn up significant hits, but considering the timespan and divergence in function this is not necessarily surprising. As discussed above, homology between the F1F0-ATP synthetase and flagellum is commonly suggested, but explicit protein-protein homologies are never proposed, and the assumption that the rotational mechanisms of the two systems are homologous implies a quite radical transformation of ATP synthetase components.� However, several recent discoveries suggest specific homologies that are much more conservative than those implied by previous workers.� First, FliH forms a (FliH)2FliI heterotrimer with FliI (Minamino and Macnab, 2000; Minamino et al., 2001).� FliH has an elongated shape (Minamino et al., 2001), and both FliI and FliH are soluble cytoplasmic components that associate intrinsically with the membrane and with lipid vesicles (Auvray et al., 2002).� If the FliH2 homodimer associates with the FliI6 complex in vivo, all of this begins to look suspiciously similar to the association (Figure 4b) between the F1F0-ATP synthetase F1-α3β3 and F0-b subunits: two elongated F0-b subunits form a dimer and interact with F1-α3β3.� In F0-b it is the N-terminal region that associates with the membrane, and the C-terminal region with the N-terminal regions of F1-α3β3 (Boyer, 1997; Weber and Senior, 2003).� In FliH it is known that the C-terminal region associates with N-terminal region of FliI (Gonzalez-Pedrajo et al., 2002), but the region responsible for membrane association is undetermined (Auvray et al., 2002); F0-b � FliH homology would predict that the FliH N-terminus associates with the membrane.� Although BLAST searches on FliH only return F0-b as a non-significant hit, a search of NCBI�s CDART (Geer et al., 2002) based on FliH does retrieve F0-b as a result with similar domain architecture (using the default e-value cutoff of 0.01), another point in favor of the hypothesis of homology.� Jackson and Plano (2000) report that the Yersinia pestis FliH homolog YscL (corresponding to SctL/HrpE in Figure 4a) has low but significant sequence similarity with the e subunit of the archaeal ATPase of Methanococcus jannaschii and the e subunit of the vacuolar ATPase of Desulfurococcus spp.; these subunits are the homologs of the b subunit of the F1F0-ATP synthetase.� Thus the present scenario predicts that careful multiple alignment of FliH sequences with bacterial F0-b and the corresponding archaeal and eukaryotic homologs (all of which would be equally related to FliH) will confirm homology.
Can further homologies between flagella and the F1F0-ATP synthetase be discerned?� In the F1F0-ATP synthetase, an F1-δ monomer associates with the proximal end of F1-α3β3 and F0-b2.� In the type III export apparatus, it is FliJ that interacts with FliI and FliH2. FliJ seems to be required for the export of all flagellar components, and so has been interpreted as a general chaperone in the cytoplasm (Macnab, 2003).� However, this observation is equally well explained if FliJ is a required part of a FliI6FliH2 complex essential for export.� Both FliJ and F1-δ have a similar size and N-terminal binding sites to the N-terminal regions of FliI/F1-α.� There may also be a structural similarity: FliJ has a high probability of exhibiting an N-terminal α-helical coiled-coil arrangement (Macnab, 2003), using sequence-based predictions (Lupas et al., 1991, method implemented at http://www.ch.embnet.org/software/COILS_form.html).� F1-δ has several conserved α-helices at its N-terminal binding site to F1 (Weber et al., 2003b).� Although predictions do not generally yield a high probability of coiled-coil structure for F1-δ, a cursory non-exhaustive sampling of orthologs shows that at least one FliJ protein does not show a high probability prediction of coiled-coil structure either (Buchnera aphidicola, accession no. P57179) while at least one F1-δ protein does (Rhodopseudomonas blastica, accession no. P05437).� It appears that the C-terminal region of F1-δ associates with the C-terminal region of F0-b2, although the details remain to be worked out (Weber and Senior, 2003).� Regarding the FliJ-FliH2 interaction, Fraser et al. (2003) favor a model where FliJ interacts with the N-terminal region of FliH2, but their data (Gonzalez-Pedrajo et al., 2002) shows that deletions in either the N-terminus (perhaps the region that associates with the membrane) or middle (dimerization region) of FliH preclude FliJ binding; thus failure of FliJ binding could be due to general malformation of FliH2 due to the failure of FliH to dimerize (middle deletion) or associate with the membrane (N-terminal deletion).� Homology between F1-δ and FliJ would predict that FliJ-FliH interaction is actually mediated through the C-terminal regions of each, but that the association may be rather weak, as it is between F0-b2 and F1-δ (Weber and Senior, 2003).�
Similarities in F1F0-δca, the integral membrane proteins FliPQR of the type III export apparatus, and the proteins SecFEY of type II secretion proteins were pointed out by Aizawa (2001), who calls these triplets the �proto-channel� and suggests homology.� His evidence is of a general nature (calculated similarities in molecular size, aliphatic index, instability index, and isoelectric point) and so cannot be accepted uncritically.� In particular, it is no longer thought that F1-δ (or its eukaryote homolog OSCP) is associated with the membrane or ATP synthetase stalk (Weber et al., 2003a), and the evidence discussed above points to a different homology for F1-δ. However, the proposed matches between FliQ--F0-c and FliR--F0-a are decent in terms of protein size and also the number of transmembrane helices of the respective proteins (Table 5).� And surprisingly, extrapolating the homology hypothesis to match the two remaining type III secretion components (FliO and FliP) to the two remaining synthetase components (F1-ε and F1-γ, respectively) also seems to provide plausible matches in terms of size.� When the similarities between F1F0-ATP synthetase and type III export components are tabulated (Table 5), it is apparent that that each component of the F1F0-ATP synthetase can be matched to a component of the type III export apparatus with a similar size and topology, as far as evidence is available (the function and structure of the flagellar proteins FliOPQR are poorly understood).�
|
Table 5: Similarities between proteins of the F1F0-ATP synthetase and the flagellar type III export apparatus that may suggest homology.� Protein size is the length in amino acids for E. coli.� TMH = Transmembrane helices.� Little detailed information on FliOPQR is available, the topologies listed are the predictions of Minamino and Macnab (1999).� Data taken from several sources: general ATP synthase component information (Boyer, 1997, updated by later references); FliI--F1-β homology (Gogarten et al., 1992; N-terminal F1-α to N-terminal F1-δ interaction (Weber et al., 2003a); FliIHJ (Minamino and Macnab, 2000; Minamino et al., 2001; Auvray et al., 2002; Minamino et al., 2002; Macnab, 2003).� The membrane-associating region of FliH is not determined (Auvray et al., 2002), but the C-terminal region interactions appear similar to the C-terminal interactions for F0-b (see text), so an N-terminal association with the membrane seems likely.
|
Individually, the cited similarities are easily attributable to chance, but together they are at least suggestive.� Although detectable sequence similarity may be too much to hope for given the already very low similarity between FliI--F1-α3β3 and FliH--F0-b, the postulated homologies would be further testable by Aizawa�s technique.� Table 5 also shows that there are some apparent dissimilarities.� Notably, while both F0-c and FliQ have 2 transmembrane helices, the loop between the helices is exposed to the cytoplasm in F0-c (Birkenhager et al., 1999), while the loop between the helices in FliQ was predicted to be periplasmic (Ohnishi et al., 1997); a reversal of this finding would support the homology hypothesis.� The weakest case for homology is between F1-ε and FliO; FliO is predicted (Ohnishi et al., 1997) to have a single transmembrane helix, while the structure of F1-ε has been solved (Wilkens and Capaldi, 1998) as a two-domain protein that binds to the stalk.� However, both proteins tolerate substantial variability; F1-ε functions with large deletions (Wilkens and Capaldi, 1998) and clear homologs of FliO have not even been identified in type III virulence systems (Gophna et al., 2003).�
The hypothesis that the entirety of a primitive F1F0-ATP synthetase may have been coopted in toto into a primitive gated pore (proto-FliF and proto-FlhA/B) is certainly provocative; it would explain at a stroke the origin of most of the type III export apparatus and provide a phylogenetically basal precursor to the flagellum even though clearly basal type III secretion systems remain undiscovered.� The complex would fit well in the FliF ring; using the stoichiometry of FlhA2FlhB2 proposed by Macnab (2003), and the equivalent stoichiometry of an ATP synthetase for the other integral membrane components, FliO1P1Q~12R1, the total number of transmembrane helices is 60, well within the approximate MS-ring capacity of about 70 transmembrane alpha-helices (Fan et al., 1997).� Fan et al. estimate <3 copies of FliR per flagellum, which is consistent with the ATP synthase homology hypothesis, but also estimate 4-5 copies for FliP, which is not, so if the ATP synthetase hypothesis is true it would be expected that the FliP finding is in error.
Macnab (1999) called the homology between FliI and F1-αβ �inexplicabl[e]�.� However, there may be a relatively simple explanation.� If the postulated homology between the ATP synthetase and type III export is correct, then the key event in the origin of type III export was the association of a primitive F1F0-ATP synthetase with a proto-FlhA or FlhB inside the proto-FliF ring, converting it from a passive to active transporter.� Since little is known about the details of the coupling of ATPase activity to protein export in Type III export, this step remains speculative. Probably motion in the synthetase was linked to a conformational change in FlhA and/or FlhB, with the proton pumping function of the synthetase lost soon afterwards.� Currently there are several documented associations between FlhAB and the rest of the type III export apparatus (Macnab, 2003).� These associations include proteins in both the �F0� and �F1� regions of the type III export apparatus. FlhA or FlhB may thus take over some of the linker role that is played by F0-b in the ATP synthetase and (on the homology hypothesis) by FliH in the type III export apparatus; this would help to explain why FliH is not absolutely required for successful construction of flagella, and FliH null mutants can be compensated by mutations in FlhA and FlhB (Minamino et al., 2003).
Other possible hypotheses for the origin of the type III export apparatus are not currently ruled out, such as the idea that much of apparatus is descended from a passive channel and that only a portion of the F1F0-ATP synthetase was coopted to power transport, or that there is an ancient, obscured homology between the various secretion systems.� Alternatives are currently disfavored because they are more complex and explain the origin of fewer components.� However, even if FliI remains the only confirmed homolog to the F1F0-ATP synthetase, general considerations indicate that the evolution of an export system is not very difficult.� A diversity of export systems of varying complexity exist, and there is a functional continuum of membrane complexes ranging from single proteins and passive pores through to active, gated export systems, indicating that there are no major evolutionary puzzles to solve.� The cataloguing and categorizing of transport proteins is already yielding insights into their origin (Saier, 2003).
The ATP synthetase homology hypothesis has the advantage of numerous testable implications for the structure and function of FliHIJOPQR.� The ATP synthetase is relatively well-understood; structures have been determined for most of the components and a number of sophisticated techniques for studying the complex as a whole have been developed.� If the homology hypothesis is correct, then similar structures would be expected for the corresponding type III export components, and many of the techniques applied to the ATP synthetase should apply to the export apparatus.� It is worth noting in passing that if a significant portion of the type III export apparatus is indeed homologous to the ATP synthetase, then it becomes fairly likely that the rotary flagellum contains within it a second rotary motor powering protein export.� This is a fairly incredible notion, but would merely be the latest in a long line of surprising discoveries yielded by the study of the flagellum.� This possibility might mean that the proto-flagellar secretion system was rotating from the start (echoing the rotation-early hypotheses of Cavalier-Smith, Goodenough, and Rizzotti), although this is not a necessary postulate for the rest of the scenario to proceed.
3.3. Type III secretion system
For the remainder, the hypothesis of a primitive type III export system will be taken for granted. This complex would have transported proteins manufactured in the cytoplasm into the periplasmic space. If secretins were already available from the type II secretion system, as they probably were given the universal distribution of type II secretion, then from the start the type III export system would have been a primitive kind of type III secretion system, as small proteins could diffuse in the periplasmic space until they found an outer membrane pore and diffused out.� Digestive proteases or antibiotic molecules are likely candidates for the secreted proteins.� Alternatively, the export system could have originally secreted proteins destined for the periplasm, and later cooption of a secretin converted the export system into a secretion system.
The association of an outer membrane channel with the type III export apparatus would improve the efficiency of secretion.� This advantage would increase as exported substrates became larger, because the peptidoglycan cell wall only allows the diffusion of globular proteins with a size less than about 50 kDa (Young, 2001); as protein size increased, diffusion would be increasingly impeded.� Once a new single-step secretion channel was available it would be possible to secrete larger proteins and proteins that would be harmful if left to wander about the periplasmic space.� These are selective forces that would favor the spread and diversification of the channel, after its origin as an efficiency-improving measure.
Outer membrane secretins have been coopted repeatedly by various versions of the secretion systems discussed above (Hueck, 1998; Thanassi, 2002; Bitter, 2003); if the type III virulence system is derived from the flagellum, it probably originated in part by replacing the flagellar L- and P-ring proteins with a secretin.� The first association of a secretin with a primitive type III export apparatus was probably mediated by the simultaneous cooption of a secretin and its outer membrane lipoprotein chaperone (Dailey and Macnab, 2002).� Both of these proteins are secreted by the type II secretion pathway.� The channel to the extracellular medium could be recruited in a single step if a mutation caused the secretin to associate with the type III export apparatus. The secretin appears to cross both the cell wall and outer membrane in the Hrp pilus, and to associate with the FliF homolog (SctJ/HrcJ; Figure 4a) in the cytoplasmic membrane (Blocker et al., 2003), so having two ring proteins (the L- and P-rings in flagella) does not appear to be a prerequisite for secretion.� Thus the double ring may have been a later addition to the system, perhaps even coinciding with the early stages of improvement of the proto-flagellum and the loss or modification of the secretin (see below).
3.4. Origin of a type III pilus
In the model, flagellin and all of the proteins of the axial structure � FlgBCFG (rod), FlgE (hook), FlgKL (adaptor), FlgD and FliD (caps), in addition to FliC (flagellin) -- are descended from a common ancestral pilin secreted from the primitive type III secretion system.� All of these proteins are placed in the axial protein family (Homma et al., 1990a; Hirano et al., 2001).� Homma et al. (1990a) put the rod, hook, and first adaptor (FlgK) proteins into a closely-related subfamily.� The divergence of the axial filament family probably occurred mostly after the origin of a functioning protoflagellum; this will be discussed in a later section.� First, the origin of the pilus must be considered.
The diversity of surface structures based on secretion systems was documented in Table 4; modern flagella retain many of these functions (Moens and Vanderleyden, 1996).� To expand on a likely function of a primitive pilus, successfully adhering to a surface can be a problem for a floating bacterium: at a low Reynolds number, the boundary layer near a surface can be a significant barrier (Vogel, 1994).� A bacterium can increase its chances of attachment by secreting adhesins with an affinity for the desired surface, ensuring successive attachment if it happens to get near a surface (e.g., the adhesins secreted by autotransporters, independent of pili (Henderson et al., 1998)).� It can increase its chances still further either by putting the adhesin at the end of a filament (e.g., the PapG adhesin located at the tip of the P pilus fibrillum (Sauer et al., 2000); the flagellar cap FliD of Pseudomonas aeruginosa doubles as an adhesin (Scharfman et al., 2001)) and/or by making the filament adhesive along its whole length, which is a common occurrence in modern bacterial flagella as well as many other surface structures (Kennedy, 1987; Moens and Vanderleyden, 1996; Fernandez and Berenguer, 2000; Giron et al., 2002).� Probably any filament, adhesins or no, will have some utility in attaching to inorganic surfaces, simply by expanding effective size and surface area available for adhesion.� Even in the absence of specific adhesins, charge, hydrophobicity, and/or van der Waals forces can be exploited for more general surface adhesion (Vogel, 1988), particularly at the small scale of bacteria.
Three hypotheses present themselves as to how the ancestral pilus originated: filament-first, cap-first, and modified filament-first.� The latter hypothesis combines the best features of the filament-first and cap-first hypotheses.
3.4.1. Filament-first hypothesis
One way that the kinds of pili described above could get their start is by simple polymerization of a surface adhesin.� The adhesin would have inherited from its ancestor the ability to bind with the outer membrane channel and with the extracellular substrate; all that would have to be added is self-binding capability.� The plausibility of this step is attested by several facts: first, structures made up of multiple copies of the same subunit are biochemically ubiquitous, and the evolution of large multimeric complexes has in many instances been traced back to simpler ancestors, e.g., AAA ATPases (Mocz and Gibbons, 2001).� Second, polymerization into a filament or tubule via mutation is a quite common event: sickle-cell hemoglobin, derived by only a single substitution from regular hemoglobin, forms not only self-assembling polymers but dynamic polymers (Mitchison, 1995).� In fact, Mitchison (1995) argues that evolution can start with just about any protein fold and produce a self-assembling polymer.�
An alternative to polymerizing an adhesin is to postulate that a gene for a pre-existing filament-forming protein was coopted by transposition of the promoter and N-terminal signal sequence of an already secreted protein.� Support for this second possibility might be found in homology between flagellin and a modern filament-forming protein.� Homology between flagellin and actin has been proposed. Harris and Elder (2002) cite an 8/13 amino acid sequence match between flagellin and actin in the N-terminal region, but this could easily be due to chance.� Novikova et al. (2000) found that flagellar filaments co-precipitate with rabbit skeletal myosin, and that flagellin and F-actin compete for myosin binding, but this might be explained by a general similarity of filaments rather than homology.� The search for flagellin-actin homology is somewhat misguided in any case, because actin is a eukaryotic protein, so ancient prokaryote actin homologs such as FtsA (Mitchison, 1995) would be more appropriate subjects.� Similarly, what should be sought is not the homolog of flagellin but the homolog of the entire axial protein family.� Given the large divergence of flagellin from the more conserved rod and hook members of the family (Homma et al., 1990a), any relationships outside of this family are bound to be difficult to detect.
A simple assumption is that the first filament was a chain of monomers, probably in an open helix. Longer filaments are presumably better for adherence than short filaments, and thus selection for adhesion can be expected to favor longer filaments.� Once a polymer filament of reasonable length has been built, however, there may be a difficulty in extending it.� The problem does not arise if filament subunits are added at the base, as occurs in type IV pili: type IV pili are based on type II secretion systems, which use a two-step process to transport proteins.� First, proteins are exported into the periplasm, and then they are pushed out through a secretin, perhaps via a �plunger�-type mechanism involving a pseudopilus (Thomas et al., 2001).� However, the type III secretion system exports proteins from the cytoplasm in one step.� By exporting a number of individual subunits, a short filament binding to the outer membrane pore can be formed, but its possible length will be severely limited by the decreasing chances of successfully adding monomers to the receding distal tip.� This problem might be overcome in a gradual manner by modifications to the open helix, so that it better corralled the monomers as they exited the secretin. Each mutation that brought the turns of the helix closer together would decrease the rate of monomer escape, and allow the extension of the filament.� A tube with closed or nearly closed walls would be the optimal solution, and selection for rigidity (necessary for very long filaments) would also favor the closed tube.�
The result would be something rather like the modern type III virulence pili, which appear to have far less complex axial structures than flagella.� Indeed, despite several investigations it has yet to be determined that the Hrp pilus has any axial components (rod-like, hook-like, etc.) apart from the main protein of the pilus, HrpA (MxiH/PrgI in Shigella/Salmonella; Blocker et al., 2003; Aizawa (2001) tentatively suggests a few others).� The extracellular portion of the filament seems to extend continuously into the secretion complex (Fernandez and Berenguer, 2000) whereas in flagella there is a distinction between filament, hook, and rod.
It might be objected at this point that the flagellum requires the cap (FliD) in order to chaperone the flagellin subunits into place at the elongating tip of the filament; without it, they diffuse away and are lost (Blocker et al., 2003).� The hook has its own temporary cap (FlgD), and it has been suggested, but not proven (Hirano et al., 2001; Berg, 2003; Macnab, 2003), that the rod has a cap protein as well (FlgJ).� However, the necessity of the cap for successfully assembling subunits is ambiguous. Flagellin will self-assemble into filaments in vitro (Hirano et al., 2001).� No cap has been identified in any type III virulence systems (Blocker et al., 2003), and although PrgJ has been suggested as a possible cap for the Salmonella needle (Sukhan et al., 2003), the evidence is indeterminate as Sukhan et al. could not detect PrgJ in sheared-off needles and did not detect it at needle tips using immunoelectron microscopy (they therefore suggest that PrgJ may be a basal component).� The polar flagellum of Vibrio grows normally without the cap (Bardy et al., 2003), probably because it is sheathed by an extension of the cell membrane (McCarter, 2001) that constrains the subunits.� Finally, even in the canonical E. coli flagellum the adaptor proteins FlgK and FlgL are added without any capping structure (Macnab, 2003), leading Macnab (2003) to argue that �capping structures are perhaps best viewed as a means of increasing efficiency of addition rather than as an absolute requirement.�� On this view, the cap could be a relatively late evolutionary addition to the pilus structure, originating by pentamerization of a pilus subunit and initially improving speed and efficiency of pilus assembly.� Later co-adaptation between filament and cap subunits would make it a more-or-less required feature.
An alternative to gradual formation of a hollow pilus would be to start with the cap.� In Pseudomonas aeruginosa, FliD serves not only as a cap protein, but also as an adhesin.� P. aeruginosa infects the human respiratory system by adhering to mucins.� FliD mutants were found to be nonadhesive, which could occur either because FliD is a necessary adhesin, or because the flagellar filament fails to assemble without the cap. However, Arora et al. (1998) found that FliC null mutants retained adhesive ability.� This implies that it is FliD specifically which serves as the adhesin, and not the whole flagellar filament, a conclusion supported by additional lines of evidence (Arora et al., 1998; Scharfman et al., 2001).� On the filament-first hypothesis, an adhesin attached to the outer membrane secretin mutated to form a polymeric chain.� The fact that caps can be adhesive, however, suggests an alternative hypothesis.� Instead of a mutation forming a polymeric chain, the mutant adhesin formed an oligomer that associated with the distal rim of the outer membrane secretin, approximately covered the mouth of the secretin, and allowing more adhesin monomers to be packed into the available space.� In this case, a pentamer ring was approximately the right size.� Once this was established, however, the utility of the adhesive �secretin cap� would be further improved if it could be extended away from the surface of the cell. This could occur by mutation of a duplicate cap protein that formed a slightly wider ring.� This ring would associate with the base of the cap, but the size mismatch would allow the insertion of more subunits, forming a short pilus in one step.� On this view, the primitive pilus would derive its structure of ~5 subunits per turn from the pentameric cap, rather than the reverse.� The channel inside the pilus would be descended from the hole in expanded ring.
3.4.3. Modified filament-first hypothesis
The filament-first hypothesis has the disadvantage of explaining the addition of distal subunits to the filament before a tube structure could evolve.� The cap-first hypothesis has the difficulty that a pentameric proto-cap covering the surface of the secretin pore might impede the secretion of other substrates.� The difficulty is not insurmountable, as secreted substrates might escape from beneath the sides of the cap, or alternatively might knock loose the cap, which is then replaced by continually secreted cap protein (both mechanisms may operate in modern flagella).� However, these problems dissolve if the hypothesized adhesin pentamer were to initially form a ring atop the secretin, instead of a cap.� Secretion of diverse substrates could then continue unabated without continual secretion of the adhesin protein.� From here, a proto-pilus could easily be formed by a mutant adhesin that polymerized the ring into a tube.� This hypothesis is simpler and more appealing because it combines the advantages of the two previous scenarios: a pilus initially assembled by simple mechanisms without a cap, but having a well-formed tube structure from the start, and allowing the uninterrupted secretion of other type III secretion system substrates.� On this hypothesis, the proto-cap would again be a late addition (a modified pilus protein) increasing assembly efficiency.�
It is interesting to reflect on the surprise many researchers felt when the mechanism of flagellar filament construction � adding subunits at the tip rather than the base � was discovered.� It has been called �astonishing� and �somewhat bizarre� (Macnab and DeRosier, 1988).� However, with the modified filament-first hypothesis in hand, the decidedly unintuitive method of filament assembly used by the flagellum can be seen as a product of the constraint of building a pilus from the starting point of type III secretion.� That a flagellum can also be built perfectly well from the base is shown by the archaeal flagellum with its type IV secretion-like system (Peabody et al., 2003).� But unlike the type IV secretion system, which has periplasmic ATPases and other outer membrane-associated active transport components, the type III system had no mechanism of powered outer membrane transport available to build on.
3.4.4. Improvements on the type III pilus
Once a primitive pilus has evolved, a number of rapid improvements can be expected.� First would be optimization of the pilin protein for its new role, under selection for increased strength, minimizing breakage, increased speed of assembly, etc.� Addition of the cap and subsequent coevolution of the pilin and cap subunits could have occurred fairly early, particularly as the pili became very long and assembly times became significant.� Pilus lengthening would be selected for because it increases reach and adhesive surface area.� That lengthening is a trivial matter of regulation is shown by various lab-produced mutants that exhibit lengthened hooks, needles, etc. due to simple mutations. Type III pili might have reached the length of flagellar filaments (10 μm) long before motility originated; flagella and Hrp pili are of comparable length (He and Jin, 2003, Figure 1). Soon the type III secretion system would become a specialized pilus-secretion apparatus, and pili would be adapted for a variety of more strenuous uses.� For example, pili might be used as stalks to elevate the bacterium above the surface, in order to better access light, a particular concentration of molecules, or escape competition from bacteria on the surface, all functions of attachment organelles today (Dyer, 2003).� Duplication and modification of the pilin protein would allow greater functional flexibility, such as adhesion to different substrates and the production of certain kinds of pili based on environmental stimuli.� One important modification that might have occurred after these trends were well along would be to strengthen the pilus attachment to the cell by extending the filament down into the secretion system to attach to the export apparatus embedded in the cytoplasmic membrane.� This would have been effected by cooption of a duplicate pilin.� The core tubule structure of the flagellar rod, hook, and filament is constructed exclusively by the N- and C-terminal domains of the axial proteins; the middle domains are placed on the outside of the tubule, and in flagellin are highly modifiable and often dispensable (Cohen-Krausz and Trachtenberg, 2003).� Therefore, the �proto-rod� probably originated by loss of the outer domains, assuming that the extracellular pilus had them for adhesion or structural purposes.� This duplication event would create the ancestors of the rod/hook subfamily and flagellin.� Initially, one cap protein could chaperone the assembly of both structures, but as they diverged, the cap protein would be duplicated as well to allow specialization on each protein, assuming that modern flagellar rods have cap proteins, as has been suggested (FlgJ; Berg, 2003; Macnab, 2003).�
It is not clear that modern type III virulence pili make use of rodlike proteins at all (the filament may simply extend all the way from the cytoplasmic export apparatus out into the extracellular space), so it is also possible that differentiation of filament and rod proteins occurred later and that attachment to the export apparatus occurred via modification of the pilin subunit.� However, the hypothesis that the duplication was early helps to explain the high divergence between flagellin and the rod/hook subfamily, as well as explaining how the filament became attached to the export apparatus instead of the outer membrane secretin (although some attachment to the secretin may have remained; see below).� Another protein, FliE, serves as the adaptor protein between FliF (the MS-ring) and FlgB (the proximal rod protein).� FliE homologs have not yet been detected in type III virulence systems, so the utility of a FliE-like adaptor in nonmotile systems is ambiguous.� Here it will be assumed that it was a relatively late, post-motility addition that strengthened the attachment between the MS-ring and the rod.� Investigation of the attachment mechanisms of modern type III pili to the secretion system may shed light on the relative likelihood of these possibilities.�
3.5. The evolution of flagella
3.5.1. The selective advantage of undirected motility
Even with a complex pilus in place, the modern flagellum could not have originated in a single step. It is hypothesized that the first, very crude motility function was random dispersal.� The function was probably not stirring or gathering more food by more rapid movement, because of the previously-discussed constraints of life at a low Reynolds number.� Dispersal, on the other hand, is both a ubiquitous adaptation in biology and rather undemanding in terms of motility.� Vogel (1994) reports that passive dispersal (i.e., unpowered dispersal by wind or current) is found in every phylum of animals and division of plants.� For creatures such as bacteria, some dispersal will occur without any adaptations whatsoever: random physical events can dislodge them from the substrate, and Brownian motion and larger-scale turbulence and flow will move them about.� Even in agar, nonflagellate bacteria without other motility systems can spread via �sliding,� motility due to colony growth requiring only the production of a surfactant to reduce friction (Brown and H�se, 2001).� However, dispersal is not always a good thing for a bacterium.� Since a bacterium existing in a location is most likely descended from a successfully reproducing bacterium also at that location, and therefore the environment is conducive to reproduction, it would be expected that the best choice for a bacterium would be to stay where it is at, rather than gambling everything on a rather literal leap into the blue.� On the other hand, this logic will rapidly break down because any environment conducive to replication will soon become filled to the brim with bacteria, at which point competition for nutrients, space, light, etc. will become severe. In such a situation there are numerous potential responses (spore formation, killing fellow bacteria by secreting antibiotics, etc.), but one of them is clearly dispersal (Dusenbery, 1998; Stoodley et al., 2002).�
The best general strategy would be for the bacterium to �decide� whether or not to disperse based on environmental cues: if life is good, stay put, but if resources are scarce, go somewhere else.� In fact, this is basically what regulates the production of flagella in modern bacteria. In E. coli, the master operon (class 1) for flagella encodes FlhC and FlhD.� These proteins activate the genes for flagellar biosynthesis in the next operon (class 2), but are repressed in high glucose conditions, where nutrients are plentiful and movement is pointless.� If conditions deteriorate, the level of cyclic AMP in the cell increases, and the expression of FlhDC is activated by cAMP and the catabolite repressor/activator protein (CAP) (Berg, 2003).� Dispersal processes and evolutionary stable dispersal strategies are tractable to mathematical and computer modeling, resulting with a large literature (e.g., Gandon and Roussett, 1999; Lebreton et al., 2000; Mathias et al., 2001; Poethke and Hovestadt, 2002), although most of it is not aimed specifically at bacteria (for exceptions see Kreft et al., 2001; Kerr et al., 2002).� All that is needed for the argument to proceed at this point is that dispersal be a widely beneficial behavior.� However, the basic dynamics of random bacterial diffusion are so simply described (Berg, 1993) that a short investigation of what kind of bacterium might be expected to evolve random dispersal is irresistible.
Even dead or otherwise nonmotile bacteria have a non-trivial diffusion coefficient: a sphere with a radius r will have a diffusion coefficient Dsphere of (Berg, 1993, eqn. 4.13):
|
| (6) |

For a dead bacterium with a diameter of 2 μm, Dsphere = 2.1x10-9 cm2/sec.� The average time t it will take to diffuse a distance x is given by (Berg, 1993, eqn. 1.10):
|
| (7) |
For the above bacterium, this means that it can be expected to passively diffuse its 2 μm diameter in ~9.3 sec, purely by Brownian motion.� However, because Brownian motion is a random walk, after each �step� the diffusing particle has an equal chance of going in any direction; the overall drift is zero.� A large number of particles placed at one location will gradually spread out in all directions by diffusion, but the mean location of the population will not change from the starting point unless additional forces come into play.� The fact that average diffusion time increases with the square of the distance means that diffusion becomes an increasing poor way to travel as the dispersal distance increases.� The 2 μm nonmotile bacterium, above, takes an average of 9 seconds to diffuse one body length, but to diffuse 100 body lengths takes not 100 times as long (15 minutes) but 10,000 times as long (26 hours). These figures are somewhat misleading as they completely ignore turbulence and flow (perfume molecules would take a month to diffuse across a room if diffusion was the only relevant process; Berg, 1993), but beneath the laminar boundary layer, very near a surface, these forces will be reduced (Vogel, 1994).
Therefore, one very crude way for a bacterium adhered to a surface to disperse is to detach its adhesins and/or adhesive pili, and let passive dispersal take place.� At least one such dispersal mechanism has been documented (Coutte et al., 2003).� If the bacterium can manage to rise substantially in the boundary layer, flow or turbulence may carry it some distance, at which point it can re-secrete adhesive pili and attach to a new surface. It will probably do better if it starts out at the top of long pilus; similar dispersal-enhancing mechanisms are well known (fruiting bodies in bacteria such as Myxococcus; Caulobacter stalks).
Under what conditions would it be advantageous to enhance the effects of Brownian motion by adding crude active motility?� Usefully, Dusenbery (1997; 1998) has derived an equation allowing the calculation of the relative utility of active and passive dispersal for organisms at a low Reynolds number.� Dusenbery assumes that the organisms are spherical and that they swim at a velocity of 10 body lengths/sec, a common value at widely varying scales (Dusenbery, 1996).� Taking into account the fact that rotational Brownian motion will keep any cell from swimming in a straight line, the ratio of the diffusion coefficient with motility (Dm) to the diffusion coefficient without it (D0) is (Dusenbery, 1997, Table 2):
|
| (8) |
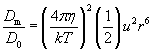
where r is again the radius and u is the cell�s swimming velocity in radii/sec (here, 20).� Dusenbery calculates that a bacterium would have to have a diameter of at least 0.64 μm in order to double its diffusion coefficient for the purpose of undirected dispersal.� Small bacteria have a very high diffusion coefficient even without motility, and for small bacteria Brownian rotation is so severe that swimming straight for any distance is impossible.� Therefore, for very small bacteria, active swimming with flagella is pointless; random Brownian motion is as good as it gets (other swimming methods may be successful, e.g. the linear motor of Spiroplasma; Trachtenberg et al., 2003).� This has the obvious implication that flagella cannot have evolved in very small bacteria; Dusenbery surveyed bacteria genera and found that the smallest motile genus had a diameter of 0.8 μm. Similar minimum size constraints were found for motility with chemotaxis, phototaxis, and thermotaxis (Dusenbery, 1997).
Equation (8) can be modified by converting the relative swimming velocity u (in radii/sec) into the absolute swimming velocity v (in cm/sec):
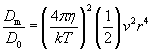 |
Equation (9) can be used to estimate the minimum swimming velocity required for a protoflagellum to substantially increase the diffusion coefficient of a cell. Dm/D0 was calculated for cells ranging from 0-8 μm in diameter, with absolute swimming velocities of 0.1, 1, and 10 μm/sec (see Figure 6).� Some advantage to diffusion would result from motility for any values of Dm/D0.� However, because the construction and movement of filaments has some cost, we have followed Dusenbery in setting the cutoff for �selectable motility function� at doubling the passive diffusion coefficient (Dm > 2D0).� As can be seen from Figure 6, Dusenbery�s result is approximately reproduced (swimming velocity here is absolute rather than relative to cell size, so slightly different input values were used): Dm/D0 is doubled for a ~0.6 μm bacterium swimming at 10 μm/sec.
|
|
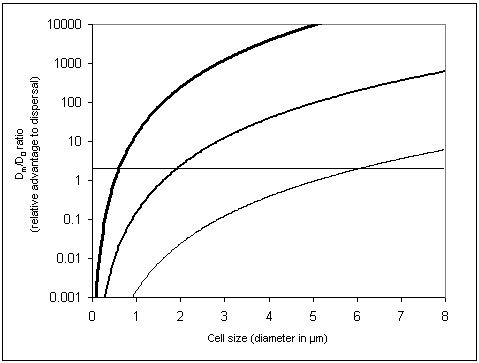
However, a crudely functioning protoflagellum cannot be expected to push a bacterium at 10 μm/sec.� The important result shown in Figure 6 is that even very slow absolute swimming velocities can result in a significant improvement in the diffusion coefficient for large bacteria.� Swimming at 1/10 the velocity of E. coli is advantageous to dispersal in a bacterium of ~2 μm diameter, and swimming at 1/100 the velocity of E. coli is advantageous to the dispersal of a bacterium with a diameter of ~6 μm.� Two factors contribute to this pattern: for larger bacteria, passive diffusion is slower, increasing the relative advantage of swimming.� Similarly, rotational diffusion is also slower for larger bacteria, but this factor enhances the efficacy of swimming as swimming runs will take longer to be randomly reoriented.�
There are additional reasons to think that the protoflagellum may have originated in a large bacterium.� Similar beneficial scaling applies if swimming velocity is considered in terms of body lengths/second: Dusenbery�s 0.64 μm bacterium has to swim at 10 body lengths/second in order to beat diffusion, but a 6 μm bacterium need only swim 0.17 body lengths/second in order to achieve a benefit.� A consideration of carbon budgets also points this direction: for a (hypothetical) very small flagellated bacterium, diameter 0.4 μm, producing 10 peritricious flagella, each ten times the body length of the cell, would cost 50% of the cell�s carbon (Mitchell, 2002).� However, for a 1 μm cell, the relative cost of 10 flagella of proportional size is only about ~2% of cell carbon.� For the 6 μm diameter cell discussed above, it is ~0.2%.� 6 μm diameter bacteria are well within the usual size range of bacteria (Dusenbery, 1997).� For a moderately large bacterium, the costs of crude, poorly functioning flagella are trivial, while the relative benefits in terms of dispersal are substantial.� The exponential nature of the relationships is such that moderate violations of the input assumptions will not greatly change the qualitative results (Dusenbery, 1997); at some moderately large size the costs of primitive motility become low and the benefits high.
The flagellar motor is made up of two proteins, MotA and MotB.� MotB binds to the peptidoglycan cell wall, allowing the complex to serve as a stator.� MotB (and perhaps MotA) also forms a proton conducting channel.� Although the exact mechanism of motor function is still mysterious, with many proposed models (Berg, 2003; Schmitt, 2003), energy from the translocation of proteins in the vicinity of MotB is somehow transformed into mechanical energy to move the rotor.� Probably this occurs by conformational change in MotA, which then binds reversibly with the rotor protein FliG, causing rotation. Speaking very metaphorically, FliG appears to act like the teeth of a gear, converting (in one model) the power stroke of MotA into rotary motion.� FliG is mounted on the central MS-ring (FliF).� Also attached to the MS-ring (perhaps mostly but not exclusively via FliG) are the switch proteins FliM and FliN.� FliM contains a receptor domain for the phosphorylated chemotaxis protein CheY-P, and the binding of CheY-P induces some kind of conformational change in FliM, FliN, and FliG that results in switching the direction of motor rotation from counterclockwise to clockwise.� This in turn results in a short �tumble� which reorients the cell, and then the flagellum returns to counterclockwise rotation.
Even given the minimal costs and substantial selective benefits of crude motility, how could the sudden origin of the rotary motor complex be mutationally possible?� The basic answer is that the ancestors of the motor proteins were already fully formed and serving other functions in the cell.� It was recently discovered (Cascales et al., 2001; Kojima and Blair, 2001) that the flagellar motor proteins MotAB have nonflagellar homologs: ExbBD and TolQR (Figure 4c).� These proteins share significant sequence similarity and all form ion channels that energize work at a distance by a third protein; ExbBD and TolQR energize outer membrane transport via action on TonB and TolA, respectively, while MotAB energize flagellar motion via action on FliG. The recently discovered homologs involved in Myxococcus gliding systems (Youderian et al., 2003) will likely add another instance, although no detailed studies of their function have been performed. The nonflagellar MotAB homologs are phylogenetically widely distributed, found in proteobacteria, cyanobacteria, Aquifex, and even in archaea (Kojima and Blair, 2001).� These facts led Kojima and Blair to note that these proteins �could perform work in contexts other than (and simpler than) the flagellar motor,� and they conclude that �ancestral forms of the MotA/MotB complex might have arisen independently of any part of the rotor.�
In order to form the motor-rotor interface, however, the origin of a third protein, FliG, must also be accounted for.� No nonflagellar homologs of FliG have been discovered (except in type III virulence systems), perhaps not surprisingly given the peculiar function of this protein and the radical change it must have undergone, whatever its ancestral function.� The structure of the middle and C-terminal domains of FliG has been resolved (Brown et al., 2002), and is primarily made of alpha helices.� Alpha helices are ubiquitous in proteins, so FliG is not necessarily structurally bizarre, despite its unusual function.� Three general possibilities present themselves for the origin of the FliG-MotAB complex.� (1) TolQR homologs were coopted via a mutation that allowed them to bind directly to FliF.� FliG was a later addition that enhanced motility by improving binding between the MS-ring and MotA, and gradually took over the interface function completely.� (2) Proto-FliG was bound to FliF before the cooption of MotAB for some other reason, perhaps a stabilization or structural function similar to that served by the FliG homologs in type III virulence systems. Mutant TolQR homologs then bound to proto-FliG.� (3) FliG was coopted simultaneously with MotAB, because it originated as a fragment of a TolA homolog that ancestrally interacted with a TolQ homolog.� The third hypothesis is the simplest and most direct pathway; the only novel interaction would be the binding of the proto-FliG to FliF; binding to the proto-MotA would be inherited.� This is less demanding than postulating the re-engineering of the interface between a TolQ homolog and its substrate (a feature of both hypotheses #1 and #2), and does not require postulating an independent cooption of FliG from an unknown source.� The hypothesis also has the advantage of being testable via determination of the structures of TonB or TolA and investigation of their interactions with ExbBD and TolQR.�
On any of the hypotheses, it is not absolutely necessary that crude motility be an immediate product; a coopted TolQRA-like complex could first have associated with the type III secretion system to enhance or help to control protein transport.� However, all that would initially be required for the very weak motility postulated above would be a slow rotation (other things being equal, a swimming velocity of 1/100th that of E. coli would imply a rotation rate similarly reduced; E. coli flagella rotate at 100 Hz, so perhaps the protoflagellum rotated at ~1 Hz).� Although the model does not hypothesize that the ancestral pilus was short, even severely truncated flagella (0.3-1.2 μm) can support residual motility (Josenhans et al., 1995; Suerbaum, 1995), lending plausibility to the notion that perfectly formed flagella are not necessary for crude motility.� In addition to the motility advantages discussed above, wiggling and twisting would probably help to dislodge the bacterium from the surface and from nearby bacteria; just such a function of certain type IV bundle-forming pili has been observed in E. coli (Knutton et al., 1999).
A possible objection here is that the ancestral pilus cannot be expected to have been freely rotating, preadapted for the addition of a motor complex; the rod might have been bound to the peptidoglycan cell wall via the P-ring, making motility impossible.� However, this assumes that a P-ring existed at this point, a dubious assumption if a secretin can bridge both the cell wall and cell membrane.� Additionally, it is actually not clear that pili are commonly rigidly fixed to the cell wall via a secretin.� It would be very interesting to know if type III and other pili, when attached to a substrate, allow a bacterium to rotate via Brownian motion in a fashion similar to motor-disabled bacteria with their flagella attached to coverslips.� Similar observations would be useful for membrane-embedded structures such as outer membrane secretins. One selective reason that might favor freely-rotating pili prior to the evolution of motility is, again, adhesion. A curved pilus that is allowed to rotate via Brownian motion can continually explore more area around a cell (particularly a large, slowly diffusing, slowly rotating cell) than a rigidly attached pilus, increasing the chances of finding a substrate.�
A final possible objection is that if the pilus was perfectly straight, then rotating it would not produce motion.� While this is true, it is irrelevant because modern pili are not always straight or stiff (Bullitt and Makowski, 1998, Figures 2-4; Honma and Nakasone, 1990; Yamashiro et al., 1994), the interconversion of pilus shapes is mutationally trivial, and there are many more ways to build a curved, helical filament than a straight one from protein subunits. Additionally, for the pilus rotated by Brownian motion postulated above, some curvature and helical character would be a requirement in order for the pilus to explore a larger area than a fixed pilus.� The exact shape of the protoflagellum is not crucial in the drag-intensive world of the low Reynolds number; Purcell (1997) has calculated that any number of peculiar rotating shapes can swim with varying efficiency (and efficiency is in fact basically energetically irrelevant at this scale; Purcell, 1977; Berg, 1993).� Purcell (1977) notes that turning anything nonsymmetrical will result in swimming.
3.5.3. Loss of outer membrane secretin
Unlike other secretion systems (type I-IV, including type III virulence systems), the type III secretion systems of bacterial flagella do not actually have an outer membrane secretin. The protein that plays the role of an outer membrane pore, FlgH, is actually a lipoprotein that Aizawa (2001) and Dailey and Macnab (2002) suggest is homologous to the Salmonella type III secretion system protein InvH.� InvH is a lipoprotein required for the insertion of the secretin InvG (SctC in Hueck�s (1998) unified nomenclature) into the outer membrane (Crago and Koronakis, 1998).� Such a mechanism is common for outer membrane secretin assembly; in the type II secretion system of Klebsiella oxytoca, the outer membrane lipoprotein PulS binds 1:1 with the secretin protein PulD, preventing periplasmic degradation and helping the localized assembly of the secretin into the outer membrane.� Twelve PulS proteins probably form a ring about the 12-PulD secretin pore (Bitter, 2003).
These observations suggest a hypothesis for the origin of the flagellar L-ring: it is not derived from an outer membrane secretin, as would be naively assumed based on its position in the outer membrane.� Rather, the FlgH L-ring may be derived from the lipoprotein that was the chaperone for the secretin of the primitive type III secretion system.� What happened to the secretin?� An obvious possibility is that it slowed the rotation of the protoflagellum.� On the present model, the primitive type III pilus originally bound to the outer membrane secretin, and later the channel was extended down to the MS-ring in the cytoplasmic membrane. However, some association or binding might have remained between the outer membrane secretin and the filament. If this was the case, it probably would not have mattered for a pili rotating by Brownian motion or rotating at low Hertz in the protoflagellum.� The outer membrane can tolerate some rotational motion of embedded components, just as membranes tolerate the lateral diffusion of proteins.� Some modern flagella are even covered by the membrane, such as the polar flagella of Vibrio, although here a special sheath evidently prevents tearing during rotation at 1500 Hz.� However, as rotation speeds increased, the risk of outer membrane tearing would increase.� A simple solution to this problem could be to delete the portion of the secretin binding to the filament; the outer lipoprotein ring would take over the role of outer membrane pore, but would not interact with the filament and would provide a bearing between the filament and outer membrane.� A channel through the cell wall would be continuously maintained if the secretin became the proto-P-ring.� Aizawa (2001) suggests homology between FlgI and the secretin InvG. The hypothesis would be strengthened if lipoproteins other than FlgH form rings in the outer membrane in other secretion systems; thus far, lipoprotein has not been found in isolated secretin complexes although the ring structure is suspected (Bitter, 2003).
For a bacterium, a sphere is the optimal shape for maximizing passive dispersal (Dusenbery, 1998).� It can thus be surmised that the cell evolving the protoflagellum was a coccus, like some flagellated bacteria today (Zaar et al., 2003).� In such a cell, the surface positioning of a flagellum is irrelevant � one place is as good as another on a sphere, so no positioning mechanism is required.� Since the model postulates that random dispersal was the original function of flagella, the first, crudely functioning protoflagellum lacked many parts that are important in modern flagella.� First, chemotaxis and switching are not required for dispersal, so neither the Che proteins nor the switch complex would have been required.� If switching is not required and the protoflagellar filament is sticking straight out at a random position on spherical cell surface, then the hook region is similarly dispensible.� Once functional motility was even marginally established, however, there would be rapid selection for improvements.� These might have occurred in more or less any order, or concurrently, so they will be discussed topically.
3.5.5. Chemotaxis and switching
The �flagellar� chemotaxis genes (Eisenbach, 2000; Table 2, this paper) are in fact not specific to flagella; the same system is coupled to diverse motility systems, including archaeal flagella and twitching motility (Faguy and Jarrell, 1999; Bardy et al., 2003). Furthermore, homologous components are tied to all manner of cellular responses to the environment; the central two-component signal transduction system (consisting of the histidine kinase CheA and the response regulator CheY in flagellar chemotaxis) is ancient, found in all three domains, and used for diverse functions.� Their evolution is discussed by Koretke et al. (2000).� Another major set of chemotaxis components, the membrane bound methyl-accepting chemotaxis proteins (MCPs) which are the receptors for attractants and repellants, have a similarly wide set of homologs with diverse functions (Zhulin et al., 2003).� More could be said about details, as there are substantial variations between the chemotaxis systems of various organisms (Eisenbach, 2000; Kirby et al., 2001), but for the purposes of this paper it will be assumed that some sort of sensory transduction system preceded the origin of the flagellum, and that one of the response regulators was the ancestor of CheY.� In modern flagella, a worsening in conditions results in the increasing phosphorylation of CheY into CheY-P.
In E. coli, switching rotation from counterclockwise (CCW) to clockwise (CW) causes the cell to tumble, reorienting it to swim in a new direction. The probability of switching is increased by the binding of CheY-P to FliM.� If concentrations of attractants are increasing during a run, CheY-P decreases and switching is suppressed, and thus favorable runs tend to last longer. If repellents are increasing, CheY-P increases, switching is promoted, and thus unfavorable runs are shortened. The cell uses this method to bias its random walk, imposing an overall drift towards regions with higher concentrations of attractants (Berg, 1993).� However, for bacteria with a diameter larger than 1.4 μm, run-and-stop or run-and-reverse strategies are more energetically favored than the run-and-tumble strategy, due to the larger costs of actively rotating a large cell (Mitchell, 2002).� As a result the run-and-tumble strategy, while common in model organisms, is far from universal.� Rhodobacter sphaeroides swims with a single, stop-start flagellum, with no reversing (Shah and Sockett, 1995; Shah et al., 2000).� Passive rotation via Brownian motion reorients the stopped cell.� This is but one of many variations on switching (Eisenbach, 2000), but probably resembles the most primitive version.
Explaining the origin of the switch complex, which couples the chemotaxis system to flagellar rotation, requires an examination of the domain structure and interactions of the switch proteins.� FliN and FliM, which make up the C-ring, are partially homologous. FliN is homologous to the C-terminal domain of FliM, and as a result the two proteins probably occupy similar positions in the C-ring, perhaps alternating in a 3 FliN:1 FliM pattern, which approximately matches their stoichiometry (Mathews et al., 1998; see also Figure 2, this paper). FliM also has a N-terminal domain with no counterpart in FliN that is the actual CheY-P receptor.� CheY-P binds to the receptor domain, increasing the probability of a switch to CW rotation (Eisenbach, 2000) via an unknown mechanism involving interactions between FliM/N and FliG (Mathews et al., 1998).� The receptor domain is homologous to the single-domain chemotaxis protein CheC of Bacillus subtilis (Kirby et al., 2001).� CheC binds reversibly to the Bacillus C-ring, and is released when it binds to CheY-P.� CheC has not been found in E. coli, but homologs are found in many early-branching bacteria, as well as archaea.� A cladogram generated for CheC and the FliM CheC-like domain shows that CheC is phylogenetically basal (Kirby et al., 2001).�
A pre-existing sensory transduction system could be coupled to flagellar rotation in a single step on the hypothesis that a FliN-like protein existed for some nonflagellar cellular response purpose, serving as a receptor for CheC.� The exact function of modern CheC is not known, but it appears to interact with CheA, CheD, and McpB, which form a receptor complex (Kirby et al., 2001).� CheC may also have a FliM-like function via interaction with the C-ring (Szurmant et al., 2003).� The ancestor of FliN might therefore be found among the other proteins that CheC interacts with.� On the model, a mutation in this FliN-like protein created a proto-FliN that bound to FliG, slowing or jamming the motor.� The reversible binding of CheC to proto-FliN, however, happened to alleviate this effect by changing the conformation of proto-FliN.� CheY-P binding to CheC would result in the dephosphorylation of CheY-P and the release of CheC from proto-FliN, resulting in the slowed-rotation behavior.� Chemotactic behavior would thereby originate by a single mutation (all other interactions would be inherited), which could then be followed by gradual improvements in the initial crude function.� This hypothesis is more economical than supposing that FliN originated for some role in structural support or enhancing export, and was later coopted to a switching function via the binding of CheC, although this remains a possibility as FliN homologs are retained in type III virulence systems for some purpose.� The first hypothesis suggests that the homolog of FliN will be found within sensory transduction systems as one of the proteins that CheC or a CheC homolog interacts with; it is difficult to know where to look with the latter hypothesis. The considerable variations in the C-ring of bacteria may yield further hints, as major variations on chemotaxis and the switch complex are known; for example, Aquifex aeolicus lacks the traditional chemotaxis system as well as FliM; Bacillus spp. have FliY (a FliM-FliN fusion protein; Bischoff and Ordal, 1992; Celandroni et al., 2000) rather than FliN. In any case, the fusion of CheC-like and FliN-like proteins would produce the FliM seen in most bacteria.
Hypothesizing a detailed pathway explaining how a stop-start switch complex could be converted into the other varieties of switching will depend on detailed knowledge of motor mechanism; many models of the flagellar motor have been proposed and the question is far from settled (Berg, 2003).� However, if proton-induced conformational changes in MotA induce some kind of power-stroke against FliG, followed by release of the FliG binding site and a return stroke to the original position, then perhaps the answer is fairly simple.� If the conformational change in the switch complex shifts the FliG binding site up or down relative to MotA, then perhaps the difference between �forward� and �reverse� �is just the difference between MotA-FliG binding on the forward power stroke or the return stroke.
Although a detailed analysis will not be performed here, the transition between random dispersal and dispersal + chemotaxis is quite gradual; adding just a small amount of directional drift to the random walk of bacteria allows gradual migration towards nutrient gradients and away from toxins or waste products. The advantages of directional drift over random diffusion are exponential (Berg, 1993), and the costs in terms of extra carbon consumption are trivial compared to the already small costs of building a flagellum in the first place.
3.5.6. Hook and additional axial components
It seems likely that the hook (FlgE) and the four rod proteins (FlgBCFG) are all duplicates of an ancestral rod protein; their sequence relationships have been described in detail elsewhere (Homma et al., 1990a; Homma et al., 1990b).� Whether phylogeny can be expected to correlate well with sequence similarity in this case is somewhat debatable, as adjacent axial components will tend to have relatively similar structural roles and signal sequences.� However, it is apparent that �adaptor� proteins (FlgK and FlgL between the hook and filament; FlgG between the hook and the rest of the rod) must have originated after the duplication of the major components; for example, as the proto-hook and proto-filament proteins began to specialize in their particular roles, the mismatches between the subunits would become increasingly troublesome, limiting further divergence.� However, duplication and modification of hook and flagellin proteins to produce adaptor proteins (a FlgE copy producing FlgK, and a FliC copy producing FlgL) would allow both tighter binding between hook and filament, and would remove the constraints on specialization of the major structures. These duplications need not have happened simultaneously; with a moderate amount of divergence, one adaptor might do (e.g., FlgG is the adaptor between the rod and hook), with a second being added as divergence continued.� This form of protein subfunctionalization (Force et al., 1999) can probably explain the rest of the axial proteins as well: the hook (FlgE) might well have originated as an adaptor between the proto-rod and proto-filament in the very early flagellum.� As FlgE specialized for the hook role, adaptors for the filament (FlgK) and rod (FlgG) would have been produced from copied hook proteins.� The reason that the flagellum has three proximal rod proteins (FlgBCF) is not clear, but may have something to do with assembly checkpoints and coordinating the addition of the P- and L-rings at the appropriate moment. FlgB is the proximal rod component, interfacing with FliF via FliE (Berg, 2003); the relative order of FlgC and FlgF has not been determined, but perhaps one assembles while the P-ring is being assembled around it, and the other assembles coincident with the L-ring.� These components are highly conserved across all known bacterial flagella, probably because of co-adapted interactions between the components, but their dispensability for building a basic filament appears to be shown by type III virulence systems, where no rod homologs have yet been discovered (Blocker et al., 2003) although the pilus protein shares similarities with axial proteins (Aizawa, 2001; Blocker et al., 2003; Cordes et al., 2003).� Thus the coordinated assembly of rod and the P- and L-rings could have been a relatively late innovation.� The origin of FliE, the adaptor between the basal rod component FlgB and the FliF MS-ring, was probably a very early event, occuring just after the origin of motility, in order to strengthen the association between the mismatched symmetries of FliF and the proto-rod.� Since FliE is exported via the type III pathway and is quite small (11 kDa), perhaps it originated as a fragment of the proto-rod protein.�
The hook capping protein FlgD probably originated as a duplicate of the putative rod capping protein (FlgJ), in a manner similar to the divergence of the rod cap and filament cap discussed previously.� However, E. coli FlgJ has a C-terminal muramidase domain in addition to an N-terminal portion that interacts with the rod proteins. This domain shows homology to other muramidases and so was probably coopted in order to speed up flagellar assembly by boring a hole through the cell wall. This was probably a late addition; even today muramidase activity is not absolutely required for successful flagellar assembly, probably because the assembling rod has a chance of finding a suitable gap in the peptidoglycan on its own (Hirano et al., 2001).� Some bacteria lack the muramidase domain (Rhodobacter) or FlgJ (gram-positive bacteria) entirely (Hirano et al., 2001).� An investigation of the assembly of the SctC ring in type III virulence systems, and similar structures in other systems, might shed light on just what the muramidase is for, as its requirement has not yet been reported for other secretion systems.� Perhaps positioning of the primitive type III pilus and protoflagellum was originally determined by the ability of the secretin to find a sufficient peptidoglycan gap to insert itself in; association of the secretin with proto-FliF brought the secretory structure together.� Having a dedicated muramidase in the modern flagellar pathway might simply enable flagella production on demand, at any predetermined spot, whether or not a sufficient hole in the peptidoglycan is already available.
The model has arrived at something like the common ancestor of all currently known bacterial flagella. Some of the variant flagella, such as those found in gram-positive bacteria, spirochetes, Aquifex, or Rhodobacter, might in fact be early offshoots of flagellar evolution. This is not required on the current model, but an improved understanding of bacterial phylogeny may change the situation.� In any case, most of these variants are probably derived (Cavalier-Smith, 2002a), as are many other minor variations that are known (Eisenbach, 2000; Bardy et al., 2003).�
4. Conclusions
The detailed evolutionary model described above is summarized in Figure 7.� The role that various evolutionary processes played in the model can now be roughly quantified.� Only one major shift of function occurred at the system level, the transition from a pilus to a protoflagellum.� All of the other changes in system function can be seen as minor modifications of a basic function; if these are enumerated (export --> secretion --> adhesion --> pilus, and dispersal --> taxis), then four minor shifts of function occurred. In all cases a �shift� in function is actually more accurately described as an addition of function at the system level, as previous functions are maintained.� At the level of subsystems (consisting of two or more proteins), the cooption events can be tabulated: subsystem cooption was invoked for the origin of the core export apparatus, outer membrane secretin (proto-FlgI) and lipoprotein chaperone (proto-FlgH), the adhesin ancestral to the axial protein family, the motor complex, and the chemotaxis/switch complex, for a total of five subsystem cooption events.� In each of these cases, cooption occurred by the mutation of one protein to link two preexisting systems (Figure 7), followed by the duplication and integration of the new subsystem proteins into the major system. Except for the major transition between pilus and motility, subsystem cooption was associated with improvements of system function rather than major changes in system function.� At the gene level, duplication events within the core system were invoked 11 times for origin of 12 axial proteins from one, and an additional time for the divergence of FliN and FliM.� None of these events requires postulating functional shift at the subsystem or system levels.� Addition of a new domain with novel functionality was identified twice (FliN+CheC --> FliM, rod cap+muramidase --> FlgJ), although it probably occurred in additional instances where homologies are currently more vague.� It appears that loss of a component is only a possibility for the outer membrane secretin of the primitive type III secretion system, although if this became FlgI then no component loss events are necessary. This is the case even though some components that are ancient on the model (e.g., FliH) are apparently not absolutely required in modern flagella (Minamino et al., 2003).� All other changes at all levels were matters of gradual improvement of function, i.e. optimization and co-adaptation of components. Even at this early stage of development, the model gives decent estimate of the relative importance of various evolutionary processes involved in the origin of complex biochemical systems.
|
|
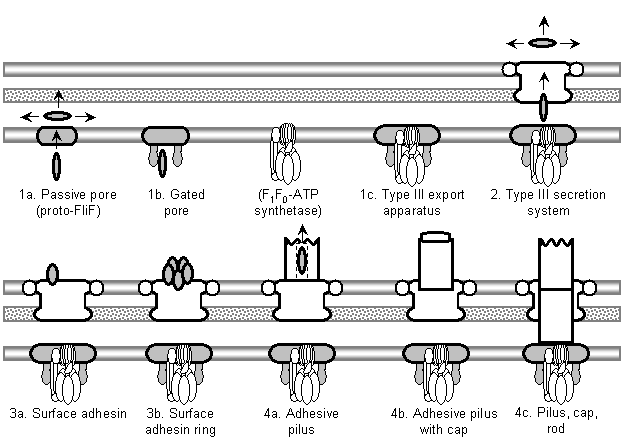
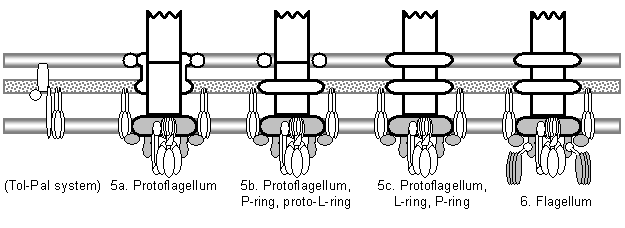
Even the present extended treatment has left out detailed discussion of the origin of the chemotaxis and regulatory proteins listed in Table 2.� However, many of these proteins have homologs functional in different systems, and the chaperones of axial proteins might have originated by duplication in a fashion similar to the axial proteins themselves. The evolution of the organization of flagellar genes and operons also deserves attention, although the precise organization found in modern bacteria is probably not essential (Kalir et al., 2001).
4.1. Evaluating the model
Biological evidence supporting the model is summarized in Table 6, in terms of extant analogs to the hypothesized intermediates and nonflagellar homologs of system components.� Of the 30 major structural components listed in Table 1, 12 are axial proteins and probably share a common (unidentified) ancestor, a hypothetical type III pilin subunit.� Of the remaining 18 components, four (FliI, MotA, MotB, and FliM) have well-accepted nonflagellar homologs based on significant sequence similarity. Suggestive evidence of homology exists for eight components, FliHJOPQR (with components of the ATP synthetase), the P-ring FlgI (with secretins), and the lipoprotein FlgH (with lipoprotein chaperones of secretins).� On the basis of interactions with other components with identified nonflagellar homologs, homologies can be postulated, with little current supporting evidence, for two components, FlgA (with other secretin-associated proteins secreted by the type II secretion system), and FliG (with a fragment of a TolA homolog). Finally, five components (FliF, FlhA, FlhB, FliN, and the ancestor of the axial proteins) have no identified potential homologs, although nonflagellar ancestral functions are not difficult to postulate.� The type III virulence system contains homologs of most of these proteins (probably including an axial protein; Cordes et al., 2003), but as discussed previously its phylogenetic position is controversial.
|
Table 6: Functions and analogs at each stage of the presented model.� See Figure 7 and text for further details.�
|
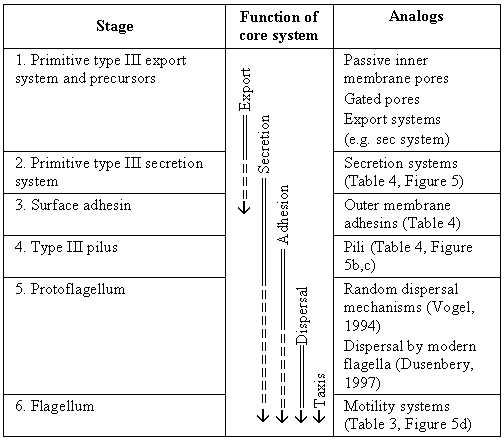
At this early stage of investigation this mixed bag should not be surprising.� Structural information (which is conserved even when sequence similarity is lost) is not available for most of the proteins, and current sampling of bacterial genomes is not very balanced.� However, the homologies postulated provide opportunities to test the model with future observations: if the model presented here is correct, then it is expected that nonflagellar homologs for most flagellar proteins will be found serving the suggested functions, in the suggested systems. Similarly, the model can be falsified by discovery of homologies in unexpected locations: for example, if the proteins of the flagellar basal body are discovered to be homologous to proteins of the junctional pore of gliding motility rather than a primitive type III secretion system, then the entire model would be overthrown and replaced by a model relating these two systems.
The proposed analogies (Table 6) provide another set of tests of the model.� Each of the systems proposed as analogies to stages in flagellar evolution is a piece of evidence that the selective forces invoked in the model are common; the fact that the functions of secretion, adhesion, pilus formation, and motility appear to be related in analogous systems lends support to the model, which postulates transitions between these functions.� The model would be weakened if the proposed analogies, mostly based on well-studied laboratory organisms, were found to actually be rare in free-living prokaryotes.� On the other hand, the discovery of further similar analogs will strengthen the model � for example, it is expected that many additional components of the archaeal flagellum will be determined to be homologous to type IV secretion (Peabody et al., 2003).� The conclusions of the simple cost-benefit model proposed here can also be tested via analogs.� Calculations indicated that the cost-benefit tradeoff is strongly in favor of motility, even very crude motility, in a moderately large bacterium.� It would therefore be expected that, in an experimental environment where dispersal is advantageous, selection would favor the retention of even severely impaired (but still motile) flagella for large bacteria, while similarly impaired flagella would be selected against in small bacteria.� Similarly, attempts to evolve crude motility in the lab (or re-evolve motility after the deletion of a crucial component) would only work if large bacteria are used.� As there are experimental conditions where it is selectively advantageous for bacteria to lose motility (Velicer et al., 2002), a careful consideration of the microbial environment would be required.�
4.2. The evolution of other microbial motility systems
The present model has several implications for the evolution of other prokaryote motility systems. The conclusions of the cost/benefit analysis, that stirring is an unlikely intermediate function and that even crude motility is advantageous for dispersal in large bacteria, will apply to the evolution of any type of flagellar-like motility in prokaryotes (the tiny Spiroplasma are apparently motile, but use a radically different system).� However, these conclusions cannot be generalized to the evolution of the eukaryotic cilium, as many eukaryotes have reached the size where stirring and swimming become useful feeding behaviors (Vogel, 1994).� Although detailed information on mechanism and homologies is not yet available, gliding motility and archaeal flagella probably both originated via evolutionary processes analogous to the present model, by cooption of pre-existing secretion systems.� This basic idea has already been proposed for archaeal flagella (Bayley and Jarrell, 1998; Peabody et al., 2003).� The fact that archaeal and bacterial flagella are completed unrelated appears to weaken Cavalier-Smith�s (2002a) argument that archaea are derived; however, if type IV secretion systems can be found in gram positive bacteria then a plausible ancestor for archaeal flagella would exist in Cavalier-Smith�s scheme.� Presumably, standard bacterial flagella could not be adapted to hyperthermophillic, hyperacidic conditions (Cavalier-Smith, 2002a), and the archaeal cenancestor was forced to re-evolve a completely new form of flagellum.
4.3. The construction of evolutionary models
It is sometimes alleged that the construction of evolutionary models amounts to nothing more than the telling of �just-so stories.�� However, the putative originators of this criticism, Gould and Lewontin (1979), only attacked scenarios that were untestable or untested.� They particularly focused their criticism of �adaptive storytelling� on cases where the adaptive function of the trait in question was highly dubious, such as human sacrifice (Gould and Lewontin, 1979).� Their point was that some traits might be explained by processes other than selection.� They never argued that systems like the bacterial flagellum, where function, complexity, and adaptiveness are obvious, might have an explanation not involving the extended action of natural selection.�
A related objection to evolutionary modeling is that it is armchair theorizing, unrelated to the practical concerns of the present day.� However, an examination of recent discoveries of nonflagellar homologs of flagellar components shows that this is not the case.� The recognition of homology between flagella and type III virulence systems has contributed greatly to an understanding of the latter, which are implicated in many diseases of humans, livestock, and crops (Hueck, 1998; Cornelis and Van Gijsegem, 2000; B�ttner and Bonas, 2002; Blocker et al., 2003; He and Jin, 2003).� Similarly, the homology between ion channels and flagellar motor proteins contributes to the understanding of the still-mysterious mechanism of the flagellar motor (Schmitt, 2003; Zhai et al., 2003).� In the case of the present model, the hypothesis of more extensive homology between the F1F0-ATP synthetase and the type III export apparatus, if true, has important implications, as the integral membrane components are the most poorly understood portion of the flagellum and type III virulence systems (Macnab, 2003).
A final advantage of constructing an evolutionary model is that it encourages the synthesis of data, relating the discoveries of specialist subfields in a coherent framework. Such a framework is a prerequisite for more detailed evolutionary investigations, providing research questions and hypotheses to test, and challenging dissenters to come up with better models.� Until now a detailed evolutionary model had never been seriously attempted for the bacterial flagellum, and even this fairly basic survey has yielded several discoveries that were not obvious at the outset.� The bacterial flagellum (and prokaryote motility systems in general) probably arose in large, coccus-shaped bacteria that were essentially modern in terms of complexity.� It is not necessary to suppose that the flagellum co-evolved with the cell wall and membranes before the last common ancestor of life.� This would be a much more difficult event to study in any case.� The previously accepted homologies between flagellar components and nonflagellar systems (such as for FliI and MotAB) are not the strange anomalies they appear to be when viewed in isolation, rather they fit well into a gradual model of flagellar evolution, and give clues as to where further homologies may be discovered.� Cooption of preexisting subsystems are the key events of interest in the model.� Gene duplications within the system primarily add complexity after the origin of the protoflagellum, and other processes, such as domain swapping and the loss of �scaffolding� components, are relatively minor players.� Finally, in light of the organized complexity and apparent �design� of the flagellum, the very fact that a step-by-step Darwinian model can be constructed that is plausible and testable significantly weakens the suggestion that extraordinary explanations might be required.
5. Acknowledgements
This work could not have been accomplished without help from numerous individuals, who supplied ideas, references, encouragement, and helpful comments.� First, Ian Musgrave, his work on this topic, and his critique of this paper were inspirational, and he was a helpful discussant.� Pete Dunkelberg, Matt Inlay, Alan Gishlick, Wesley Elsberry, John Wilkins, Pim van Meurs, and many others gave help in terms of editing and informal discussions.� The initial inspiration was another paper, on the evolution of biological complexity, written for a course taught Jim Proctor, although the present paper ended up going far beyond the original topic.
6. References
Aizawa, S. I., 2001. Bacterial flagella and type III secretion systems. FEMS Microbiol Lett. 202 (2), 157-164. DOI link.
Albertini, A. M., Caramori, T., Crabb, W. D., Scoffone, F. and Galizzi, A., 1991. The flaA locus of Bacillus subtilis is part of a large operon coding for flagellar structures, motility functions, and an ATPase-like polypeptide. J Bacteriol. 173 (11), 3573-3579.
Anandarajah, K., Kiefer, P. M., Donohoe, B. S. and Copley, S. D., 2000. Recruitment of a double bond isomerase to serve as a reductive dehalogenase during biodegradation of pentachlorophenol. Biochemistry. 39 (18), 5303-5311. DOI link.
Arora, S. K., Ritchings, B. W., Almira, E. C., Lory, S. and Ramphal, R., 1998. The Pseudomonas aeruginosa flagellar cap protein, FliD, is responsible for mucin adhesion. Infect Immun. 66 (3), 1000-1007.
Auvray, F., Ozin, A. J., Claret, L. and Hughes, C., 2002. Intrinsic membrane targeting of the flagellar export ATPase FliI: Interaction with acidic phospholipids and FliH. Journal of Molecular Biology. 318 (4), 941-950. DOI link.
Bardy, S. L., Ng, S. Y. and Jarrell, K. F., 2003. Prokaryotic motility structures. Microbiology. 149 (Pt 2), 295-304. DOI link.
Bayley, D. P. and Jarrell, K. F., 1998. Further evidence to suggest that archaeal flagella are related to bacterial type IV pili. J Mol Evol. 46 (3), 370-373.
Berg, H. C., 1993. Random Walks in Biology. Princeton University Press, Princeton.
Berg, H. C., 1998. Keeping up with the F1-ATPase. Nature. 394 (6691), 324-325. DOI link.
Berg, H. C., 2003. The rotary motor of bacterial flagella. Annu Rev Biochem. 72, 19-54. DOI link.
Berg, H. C. and Anderson, R. A., 1973. Bacteria swim by rotating their flagellar filaments. Nature. 245 (5425), 380-382.
Berry, R. M., 2000. Theories of rotary motors. Philos Trans R Soc Lond B Biol Sci. 355 (1396), 503-509. DOI link.
Birkenhager, R., Greie, J.-C., Altendorf, K. and Deckers-Hebestreit, G., 1999. F0 complex of the Escherichia coli ATP synthase . Not all monomers of the subunit c oligomer are involved in F1 interaction. Eur J Biochem. 264 (2), 385-396. DOI link.
Bischoff, D. S. and Ordal, G. W., 1992. Identification and characterization of FliY, a novel component of the Bacillus subtilis flagellar switch complex. Mol Microbiol. 6 (18), 2715-2723.
Bitter, W., 2003. Secretins of Pseudomonas aeruginosa: large holes in the outer membrane. Arch Microbiology. 179 (5), 307-314. DOI link.
Block, S. M., 1997. Real engines of creation. Nature. 386 (6622), 217-219. DOI link.
Blocker, A., Komoriya, K. and Aizawa, S. I., 2003. Type III secretion systems and bacterial flagella: Insights into their function from structural similarities. Proc Natl Acad Sci U S A. 100 (6), 3027-3030. DOI link.
Boyer, P. D., 1997. The ATP synthase - A splendid molecular machine. Annu Rev Biochem. 66, 717-749. DOI link.
Broome-Smith, J. K. and Mitsopoulos, C., 1999. Overview: Transport of molecules across microbial membranes -- a sticky situation to get to grips with, in: Broome-Smith, J. K., Baumberg, S., Stirling, C. J. and Ward, F. B. (Eds.), Transport of molecules across microbial membranes, General Society for Microbiology, Cambridge University Press, Cambridge, UK, pp.1-14.
Brown, I. I. and H�se, C. C., 2001. Flagellum-independent surface migration of Vibrio cholerae and Escherichia coli. J Bacteriol. 183 (12), 3784-3790. DOI link.
Brown, P. N., Hill, C. P. and Blair, D. F., 2002. Crystal structure of the middle and C-terminal domains of the flagellar rotor protein FliG. Embo J. 21 (13), 3225-3234. DOI link.
Buchanan, S. K., 2001. Type I secretion and multidrug efflux: transport through the TolC channel-tunnel. Trends in Biochemical Sciences. 26 (1), 3-6. DOI link.
Bullitt, E. and Makowski, L., 1998. Bacterial adhesion pili are heterologous assemblies of similar subunits. Biophys J. 74 (1), 623-632.
B�ttner, D. and Bonas, U., 2002. Port of entry � the type III secretion translocon. Trends Microbiol. 10 (4), 186-192. DOI link.
Campbell, N. A., 1993. Biology. Benjamin/Cummings, Redwood City, CA.
Campos-Garcia, J., Najera, R., Camarena, L. and Soberon-Chavez, G., 2000. The Pseudomonas aeruginosa motR gene involved in regulation of bacterial motility. FEMS Microbiol Lett. 184 (1), 57-62. DOI link.
Cao, T. B. and Saier, M. H., Jr., 2003. The general protein secretory pathway: phylogenetic analyses leading to evolutionary conclusions. Biochim Biophys Acta. 1609 (1), 115-125. DOI link.
Capaldi, R. A. and Aggeler, R., 2002. Mechanism of the F1-F0-type ATP synthase, a biological rotary motor. Trends in the Biochemical Sciences. 27 (3), 154-160. DOI link.
Cascales, E., Lloubes, R. and Sturgis, J. N., 2001. The TolQ-TolR proteins energize TolA and share homologies with the flagellar motor proteins MotA-MotB. Mol Microbiol. 42 (3), 795-807. DOI link.
Cavalier-Smith, T., 1978. The evolutionary origin and phylogeny of microtubules, mitotic spindles and eukaryote flagella. Biosystems. 10 (1-2), 93-114.
Cavalier-Smith, T., 1982. The evolutionary origin and phylogeny of eukaryote flagella. Symposia of the Society for Experimental Biology. 35 (5896), 465-493.
Cavalier-Smith, T., 1987a. The origin of cells: a symbiosis between genes, catalysts, and membranes. Cold Spring Harbor Symposia on Quantitative Biology. 52 (6111), 805-824.
Cavalier-Smith, T., 1987b. The origin of eukaryotic and archaebacterial cells. Annals of the New York Academy of Sciences. 503, 17-54.
Cavalier-Smith, T., 2001a. Obcells as proto-organisms: membrane heredity, lithophosphorylation, and the origins of the genetic code, the first cells, and photosynthesis. Journal of Molecular Evolution. 53 (4-5), 555-595. DOI link.
Cavalier-Smith, T., 2001b. Early evolution: from the appearance of the first cell to the first modern organisms (review). Quarterly Review of Biology. 76 (2), 233.
Cavalier-Smith, T., 2002a. The neomuran origin of archaebacteria, the negibacterial root of the universal tree and bacterial megaclassification. Int J Syst Evol Microbiol. 52, 7-76.
Cavalier-Smith, T., 2002b. The phagotrophic origin of eukaryotes and phylogenetic classification of Protozoa. Int J Syst Evol Microbiol. 52, 297-354.
Cavalier-Smith, T., 2002c. Origins of the machinery of recombination and sex. Heredity. 88, 125-141. DOI link.
Celandroni, F., Ghelardi, E., Pastore, M., Lupetti, A., Kolsto, A. B. and Senesi, S., 2000. Characterization of the chemotaxis fliY and cheA genes in Bacillus cereus. FEMS Microbiol Lett. 190 (2), 247-253. DOI link.
Chapman, M. R., Robinson, L. S., Pinkner, J. S., Roth, R., Heuser, J., Hammar, M., Normark, S. and Hultgren, S. J., 2002. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. 295 (5556), 851-855. DOI link.
Chothia, C., Gough, J., Vogel, C. and Teichmann, S. A., 2003. Evolution of the protein repertoire. Science. 300 (5626), 1701-1703. DOI link.
Christie, P. J., 2001. Type IV secretion: intercellular transfer of macromolecules by systems ancestrally related to conjugation machines. Mol Microbiol. 40 (2), 294-305. DOI link.
Christie, P. J. and Vogel, J. P., 2000. Bacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol. 8 (8), 354-360. DOI link.
Claret, L., Calder, S. R., Higgins, M. and Hughes, C., 2003. Oligomerization and activation of the FliI ATPase central to bacterial flagellum assembly. Mol Microbiol. 48 (5), 1349-1355. DOI link.
Cohen-Krausz, S. and Trachtenberg, S., 2003. The structure of the helically perturbed flagellar filament of Pseudomonas rhodos: implications for the absence of the outer domain in other complex flagellins and for the flexibility of the radial spokes. Mol Microbiol. 48 (5), 1305-1316. DOI link.
Copley, S. D., 2000. Evolution of a metabolic pathway for degradation of a toxic xenobiotic: the patchwork approach. Trends in Biochemical Sciences. V25 (N6), 261-265. DOI link.
Cordes, F. S., Komoriya, K., Larquet, E., Yang, S., Egelman, E. H., Blocker, A. and Lea, S. M., 2003. Helical structure of the needle of the type III secretion system of Shigella flexneri. J Biol Chem. 278 (19), 17103-17107. DOI link.
Corliss, J. O., 1980. Objection to "undulipodium" as an inappropriate and unnecessary term. Biosystems. 12 (1-2), 109-110.
Cornelis, G. R. and Van Gijsegem, F., 2000. Assembly and function of type III secretory systems. Annu Rev Microbiol. 54, 735-774. DOI link.
Coutte, L., Alonso, S., Reveneau, N., Willery, E., Quatannens, B., Locht, C. and Jacob-Dubuisson, F., 2003. Role of adhesin release for mucosal colonization by a bacterial pathogen. J Exp Med. 197 (6), 735-742. DOI link.
Cox, G. B., Jans, D. A., Fimmel, A. L., Gibson, F. and Hatch, L., 1984. Hypothesis. The mechanism of ATP synthase. Conformational change by rotation of the beta-subunit. Biochim Biophys Acta. 768 (3-4), 201-208.
Crago, A. M. and Koronakis, V., 1998. Salmonella InvG forms a ring-like multimer that requires the InvH lipoprotein for outer membrane localization. Mol Microbiol. 30 (1), 47-56. DOI link.
Dailey, F. E. and Macnab, R. M., 2002. Effects of lipoprotein biogenesis mutations on flagellar assembly in Salmonella. J Bacteriol. 184 (3), 771-776. DOI link.
Darwin, C., 1851. A Monograph of the Sub-class Cirripedia, with Figures of all the Species. The Lepadidae; or, Pedunculated Cirripedes. Ray Society, London. Online text.
Darwin, C., 1854. A Monograph of the Sub-class Cirripedia, with Figures of all the Species. The Balanidae (or Sessile Cirripedes); the Verrucidae, etc. Ray Society, London. Online text.
Darwin, C., 1862. On the various contrivances by which orchids are fertilised. J. Murray, London. Online text.
Darwin, C., 1872. The origin of species by means of natural selection. 6th edition. John Murray, London. Online text.
de Duve, C., 1995. Vital Dust: Life as a cosmic imperative. Basic Books, New York.
de Souza, M. L., Seffernick, J., Martinez, B., Sadowsky, M. J. and Wackett, L. P., 1998. The atrazine catabolism genes atzABC are widespread and highly conserved. J Bacteriol. 180 (7), 1951-1954.
DeLong, E. F. and Pace, N. R., 2001. Environmental diversity of bacteria and archaea. Systematic Biology. 50 (4), 470-478. DOI link.
DeRosier, D. J., 1998. The turn of the screw: the bacterial flagellar motor. Cell. 93 (1), 17-20.
Dillon, R., Fauci, L. and Gaver, D., 3rd, 1995. A microscale model of bacterial swimming, chemotaxis and substrate transport. J Theor Biol. 177 (4), 325-340. DOI link.
Doolittle, R. F. and Feng, D. F., 1987. Reconstructing the evolution of vertebrate blood coagulation from a consideration of the amino acid sequences of clotting proteins. Cold Spring Harbor Symposia on Quantitative Biology: The Evolution of Catalytic Function. LII, 869-874.
Durand, E., Bernadac, A., Ball, G., Lazdunski, A., Sturgis, J. N. and Filloux, A., 2003. Type II protein secretion in Pseudomonas aeruginosa: the pseudopilus is a multifibrillar and adhesive structure. J Bacteriol. 185 (9), 2749-2758. DOI link.
Dusenbery, D. B., 1996. Life At Small Scale: The Behavior of Microbes. Scientific American Library, New York.
Dusenbery, D. B., 1997. Minimum size limit for useful locomotion by free-swimming microbes. Proc Natl Acad Sci U S A. 94 (20), 10949-10954. DOI link.
Dusenbery, D. B., 1998. Fitness landscapes for effects of shape on chemotaxis and other behaviors of bacteria. J Bacteriol. 180 (22), 5978-5983.
Dyer, B. D., 2003. A Field Guide to Bacteria. Cornell University Press, Ithaca.
Eisenbach, M., 2000. Bacterial chemotaxis, Nature Encyclopedia of Life Sciences, Nature Publishing Group, London. DOI link.
Emerson, S. U., Tokuyasu, K. and Simon, M. I., 1970. Bacterial flagella: polarity of elongation. Science. 169 (941), 190-192.
Faguy, D. M. and Jarrell, K. F., 1999. A twisted tale: the origin and evolution of motility and chemotaxis in prokaryotes. Microbiology. 145 (Pt 2), 279-281.
Faguy, D. M., Jarrell, K. F., Kuzio, J. and Kalmokoff, M. L., 1994. Molecular analysis of archael flagellins: similarity to the type IV pilin-transport superfamily widespread in bacteria. Can J Microbiol. 40 (1), 67-71.
Fan, F., Ohnishi, K., Francis, N. R. and Macnab, R. M., 1997. The FliP and FliR proteins of Salmonella typhimurium, putative components of the type III flagellar export apparatus, are located in the flagellar basal body. Mol Microbiol. 26 (5), 1035-1046.
Fernandez, L. A. and Berenguer, J., 2000. Secretion and assembly of regular surface structures in Gram-negative bacteria. FEMS Microbiol Rev. 24 (1), 21-44. DOI link.
Force, A., Lynch, M., Pickett, F. B., Amores, A., Yan, Y. L. and Postlethwait, J., 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 151 (4), 1531-1545.
Francis, N. R., Sosinsky, G. E., Thomas, D. and DeRosier, D. J., 1994. Isolation, characterization and structure of bacterial flagellar motors containing the switch complex. J Mol Biol. 235 (4), 1261-1270. DOI link.
Fraser, G. M., Gonzalez-Pedrajo, B., Tame, J. R. and Macnab, R. M., 2003. Interactions of FliJ with the Salmonella Type III Flagellar Export Apparatus. J Bacteriol. 185 (18), 5546-5554. DOI link.
Gandon, S. and Roussett, F., 1999. Evolution of stepping-stone dispersal rates. Proc R Soc Lond B Biol Sci. 266 (1437), 2507-2513.
Ganfornina, M. D. and Sanchez, D., 1999. Generation of evolutionary novelty by functional shift. Bioessays. V21 (N5), 432-439.
Geer, L. Y., Domrachev, M., Lipman, D. J. and Bryant, S. H., 2002. CDART: protein homology by domain architecture. Genome Res. 12 (10), 1619-1623. DOI link.
Giron, J. A., Torres, A. G., Freer, E. and Kaper, J. B., 2002. The flagella of enteropathogenic Escherichia coli mediate adherence to epithelial cells. Mol Microbiol. 44 (2), 361-379. DOI link.
Gogarten, J. P. and Kibak, H., 1992. The bioenergetics of the last common ancestor and the origin of the eukaryotic endomembrane system. Biosystems. 28 (1-3), 131-153.
Gogarten, J. P., Kibak, H., Dittrich, P., Taiz, L., Bowman, E. J., Bowman, B. J., Manolson, M. F., Poole, R. J., Date, T. and Oshima, T., 1989. Evolution of the vacuolar H+-ATPase: implications for the origin of eukaryotes. Proc Natl Acad Sci U S A. 86 (17), 6661-6665.
Gogarten, J. P., Starke, T., Kibak, H., Fishman, J. and Taiz, L., 1992. Evolution and isoforms of V-ATPase subunits. J Exp Biol. 172, 137-147.
Gonzalez-Pedrajo, B., Fraser, G. M., Minamino, T. and Macnab, R. M., 2002. Molecular dissection of Salmonella FliH, a regulator of the ATPase FliI and the type III flagellar protein export pathway. Mol Microbiol. 45 (4), 967-982. DOI link.
Goodenough, U., 1998. The Sacred Depths of Nature. Oxford University Press, New York.
Goodenough, U., 2002. Of flagella and outboard motors, accessed online. URL: http://www.metanexus.net/archives/printerfriendly.asp?archiveid=7143.
Gophna, U., Ron, E. Z. and Graur, D., 2003. Bacterial type III secretion systems are ancient and evolved by multiple horizontal-transfer events. Gene. 312, 151-163. DOI link.
Gould, S. J. and Lewontin, R. C., 1979. The spandrels of San Marco and the Panglossian paradigm: A critique of the adaptationist programmme. Proceedings of the Royal Society of London, Series B. 205 (1161), 581-598.
Guerrero, R., Esteve, I., Pedr�s-Ali�, C. and Gaju, N., 1987. Predatory bacteria in prokaryotic communities. Annals of the New York Academy of Sciences. 503, 238-250.
Hanumanthaiah, R., Day, K. and Jagadeeswaran, P., 2002. Comprehensive analysis of blood coagulation pathways in Teleostei: evolution of coagulation factor genes and identification of zebrafish factor VIIi. Blood Cells, Molecules, and Diseases. 29 (1), 57-68.
Harris, R. J. and Elder, D., 2002. Actin and flagellin may have an N-terminal relationship. J Mol Evol. 54 (2), 283-284. DOI link.
Harshey, R. M. and Toguchi, A., 1996. Spinning tails: homologies among bacterial flagellar systems. Trends Microbiol. 4 (6), 226-231. DOI link.
Hayward, C. A., 2000. Microorganisms, Nature Encyclopedia of Life Sciences, Nature Publishing Group, London. DOI link.
He, S. Y., 1998. Type III protein secretion in plant and animal pathogenic bacteria. Annual Reviews in Phytopathology. 36, 363-392. DOI link.
He, S. Y. and Jin, Q., 2003. The Hrp pilus: learning from flagella. Curr Opin Microbiol. 6 (1), 15-19. DOI link.
Henderson, I. R., Navarro-Garcia, F. and Nataro, J. P., 1998. The great escape: structure and function of the autotransporter proteins. Trends Microbiol. 6 (9), 370-378. DOI link.
Hirano, T., Minamino, T. and Macnab, R. M., 2001. The role in flagellar rod assembly of the N-terminal domain of Salmonella FlgJ, a flagellum-specific muramidase. J Mol Biol. 312 (2), 359-369. DOI link.
Homma, M., DeRosier, D. J. and Macnab, R. M., 1990a. Flagellar hook and hook-associated proteins of Salmonella typhimurium and their relationship to other axial components of the flagellum. J Mol Biol. 213 (4), 819-832.
Homma, M., Kutsukake, K., Hasebe, M., Iino, T. and Macnab, R. M., 1990b. FlgB, FlgC, FlgF and FlgG. A family of structurally related proteins in the flagellar basal body of Salmonella typhimurium. J Mol Biol. 211 (2), 465-477.
Honma, Y. and Nakasone, N., 1990. Pili of Aeromonas hydrophila: purification, characterization, and biological role. Microbiol Immunol. 34 (2), 83-98.
Hooper, S. D. and Berg, O. G., 2003. On the nature of gene innovation: duplication patterns in microbial genomes. Mol Biol Evol. 20 (6), 945-954. DOI link.
Hueck, C. J., 1998. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 62 (2), 379-433.
Jackson, M. W. and Plano, G. V., 2000. Interactions between type III secretion apparatus components from Yersinia pestis detected using the yeast two-hybrid system. FEMS Microbiol Lett. 186 (1), 85-90. DOI link.
Jarrell, K. F., Bayley, D. P., Correia, J. D. and Thomas, N. A., 1999. Recent excitement about the archaea. Bioscience. 49 (7), 530-541.
Jarrell, K. F., Bayley, D. P., Correia, J. D. and Thomas, N. A., 2000. Archaeal flagella, Nature Encyclopedia of Life Sciences, Nature Publishing Group, London. DOI link.
Jarrell, K. F., Bayley, D. P. and Kostyukova, A. S., 1996. The archaeal flagellum: a unique motility structure. J Bacteriol. 178 (17), 5057-5064.
Jiang, Y. and Doolittle, R. F., 2003. The evolution of vertebrate blood coagulation as viewed from a comparison of puffer fish and sea squirt genomes. Proc Natl Acad Sci U S A. 100 (13), 7527�7532. DOI link.
Johnson, G. R., Jain, R. K. and Spain, J. C., 2002. Origins of the 2,4-dinitrotoluene pathway. Journal of Bacteriology. 184 (15), 4219-4232. DOI link.
Jones, W. J., Nagle, D. P., Jr. and Whitman, W. B., 1987. Methanogens and the diversity of archaebacteria. Microbiol Rev. 51 (1), 135-177.
Josenhans, C., Labigne, A. and Suerbaum, S., 1995. Comparative ultrastructural and functional studies of Helicobacter pylori and Helicobacter mustelae flagellin mutants: both flagellin subunits, FlaA and FlaB, are necessary for full motility in Helicobacter species. J Bacteriol. 177 (11), 3010-3020.
Kalir, S., McClure, J., Pabbaraju, K., Southward, C., Ronen, M., Leibler, S., Surette, M. G. and Alon, U., 2001. Ordering genes in a flagella pathway by analysis of expression kinetics from living bacteria. Science. 292 (5524), 2080-2083. DOI link.
Karner, M. B., DeLong, E. F. and Karl, D. M., 2001. Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature. 409 (6819), 507-510. DOI link.
Kennedy, M. J., 1987. Role of motility, chemotaxis, and adhesion in microbial ecology. Annals of the New York Academy of Sciences. 503, 260-273.
Kerr, B., Riley, M. A., Feldman, M. W. and Bohannan, B. J., 2002. Local dispersal promotes biodiversity in a real-life game of rock-paper-scissors. Nature. 418 (6894), 171-174. DOI link.
Khan, S., 1997. Rotary chemiosmotic machines. Biochim Biophys Acta. 1322 (2-3), 86-105.
Kim, J. F., 2001. Revisiting the chlamydial type III protein secretion system: clues to the origin of type III protein secretion. Trends Genet. 17 (2), 65-69. DOI link.
Kirby, J. R., Kristich, C. J., Saulmon, M. M., Zimmer, M. A., Garrity, L. F., Zhulin, I. B. and Ordal, G. W., 2001. CheC is related to the family of flagellar switch proteins and acts independently from CheD to control chemotaxis in Bacillus subtilis. Mol Microbiol. 42 (3), 573-585. DOI link.
Knutton, S., Shaw, R. K., Anantha, R. P., Donnenberg, M. S. and Zorgani, A. A., 1999. The type IV bundle-forming pilus of enteropathogenic Escherichia coli undergoes dramatic alterations in structure associated with bacterial adherence, aggregation and dispersal. Mol Microbiol. 33 (3), 499-509. DOI link.
Koch, A. L., 2003. Were Gram-positive rods the first bacteria? Trends Microbiol. 11 (4), 166-170. DOI link.
Kojima, S. and Blair, D. F., 2001. Conformational change in the stator of the bacterial flagellar motor. Biochemistry. 40 (43), 13041-13050. DOI link.
Koretke, K. K., Lupas, A. N., Warren, P. V., Rosenberg, M. and Brown, J. R., 2000. Evolution of two-component signal transduction. Mol Biol Evol. 17 (12), 1956-1970.
Kreft, J. U., Picioreanu, C., Wimpenny, J. W. and van Loosdrecht, M. C., 2001. Individual-based modelling of biofilms. Microbiology. 147 (Pt 11), 2897-2912.
Lebreton, J., Khaladi, M. and Grosbois, V., 2000. An explicit approach to evolutionarily stable dispersal strategies: no cost of dispersal. Math Biosci. 165 (2), 163-176. DOI link.
Lenski, R. E., Ofria, C., Pennock, R. T. and Adami, C., 2003. The evolutionary origin of complex features. Nature. 423 (6936), 139-144. DOI link.
Long, M., 2001. Evolution of novel genes. Curr Opin Genet Dev. 11 (6), 673-680. DOI link.
Lupas, A., Van Dyke, M. and Stock, J., 1991. Predicting coiled coils from protein sequences. Science. 252 (5010), 1162-1164.
Macnab, R. M., 1978. Bacterial motility and chemotaxis: the molecular biology of a behavioral system. CRC Crit Rev Biochem. 5 (4), 291-341.
Macnab, R. M., 1999. The bacterial flagellum: reversible rotary propellor and type III export apparatus. J Bacteriol. 181 (23), 7149-7153.
Macnab, R. M., 2003. How bacteria assemble flagella. Annu Rev Microbiol. 57, 77-100. DOI link.
Macnab, R. M. and DeRosier, D. J., 1988. Bacterial flagellar structure and function. Can J Microbiol. 34 (4), 442-451.
Manson, M. D., Tedesco, P., Berg, H. C., Harold, F. M. and Van der Drift, C., 1977. A protonmotive force drives bacterial flagella. Proc Natl Acad Sci U S A. 74 (7), 3060-3064.
Margulis, L., 1980. Undulipodia, flagella and cilia. Biosystems. 12 (1-2), 105-108.
Marie, C., Broughton, W. J. and Deakin, W. J., 2001. Rhizobium type III secretion systems: legume charmers or alarmers? Curr Opin Plant Biol. 4 (4), 336-342. DOI link.
Mathews, M. A., Tang, H. L. and Blair, D. F., 1998. Domain analysis of the FliM protein of Escherichia coli. J Bacteriol. 180 (21), 5580-5590.
Mathias, A., Kisdi, E. and Olivieri, I., 2001. Divergent evolution of dispersal in a heterogeneous landscape. Evolution Int J Org Evolution. 55 (2), 246-259.
Maynard Smith, J., 1975. The Theory of Evolution. Cambridge University Press.
Mayr, E., 1976. The emergence of evolutionary novelties, Evolution and the diversity of life, Harvard University Press, Cambridge, pp. 88-113.
McBride, M. J., 2001. Bacterial gliding motility: multiple mechanisms for cell movement over surfaces. Annu Rev Microbiol. 55, 49-75. DOI link.
McCarter, L. L., 2001. Polar flagellar motility of the Vibrionaceae. Microbiol Mol Biol Rev. 65 (3), 445-462, table of contents. DOI link.
Mecsas, J. J. and Strauss, E. J., 1996. Molecular mechanisms of bacterial virulence: type III secretion and pathogenicity islands. Emerg Infect Dis. 2 (4), 270-288.
Melendez-Hevia, E., Waddell, T. G. and Cascante, M., 1996. The puzzle of the Krebs citric acid cycle: assembling the pieces of chemically feasible reactions, and opportunism in the design of metabolic pathways during evolution. J Mol Evol. 43 (3), 293-303.
Merz, A. J. and Forest, K. T., 2002. Bacterial surface motility: slime trails, grappling hooks and nozzles. Curr Biol. 12 (8), R297-303. DOI link.
Miller, K. R., 2003. Answering the biochemical argument from design, in: Manson, N. (Ed.), God and design: the teleological argument and modern science, Routledge. URL: http://www.millerandlevine.com/km/evol/design1/article.html
Miller, K. R., 2004. The Flagellum Unspun: The Collapse of "Irreducible Complexity", in: Ruse, M. and Dembski, W. (Eds.), Debating Design: from Darwin to DNA, Cambridge University Press, Cambridge, MA. URL: http://www.millerandlevine.com/km/evol/design2/article.html
Minamino, T., Gonzalez-Pedrajo, B., Kihara, M., Namba, K. and Macnab, R. M., 2003. The ATPase FliI can interact with the type III flagellar protein export apparatus in the absence of its regulator, FliH. J Bacteriol. 185 (13), 3983-3988.
Minamino, T., Gonzalez-Pedrajo, B., Oosawa, K., Namba, K. and Macnab, R. M., 2002. Structural properties of FliH, an ATPase regulatory component of the Salmonella type III flagellar export apparatus. J Mol Biol. 322 (2), 281-290. DOI link.
Minamino, T. and Macnab, R. M., 1999. Components of the Salmonella flagellar export apparatus and classification of export substrates. J Bacteriol. 181 (5), 1388-1394.
Minamino, T. and Macnab, R. M., 2000. FliH, a soluble component of the type III flagellar export apparatus of Salmonella, forms a complex with FliI and inhibits its ATPase activity. Mol Microbiol. 37 (6), 1494-1503. DOI link.
Minamino, T., Tame, J. R., Namba, K. and Macnab, R. M., 2001. Proteolytic analysis of the FliH/FliI complex, the ATPase component of the type III flagellar export apparatus of Salmonella. J Mol Biol. 312 (5), 1027-1036. DOI link.
Mitchell, J. G., 2002. The energetics and scaling of search strategies in bacteria. The American Naturalist. 160 (6), 727-740. DOI link.
Mitchell, P., 1985. Molecular mechanics of protonmotive F0F1 ATPases. Rolling well and turnstile hypothesis. FEBS Lett. 182 (1), 1-7.
Mitchison, T. J., 1995. Evolution of a dynamic cytoskeleton. Philos Trans R Soc Lond B Biol Sci. 349 (1329), 299-304.
Mivart, S. G., 1871. Genesis of Species. Macmillan, London.
Mocz, G. and Gibbons, I. R., 2001. Model for the motor component of dynein heavy chain based on homology to the AAA family of oligomeric ATPases. Structure (Camb). 9 (2), 93-103. DOI link.
Moens, S. and Vanderleyden, J., 1996. Functions of bacterial flagella. Crit Rev Microbiol. 22 (2), 67-100.
Mortlock, R. P. (Ed.), 1992. The Evolution of Metabolic Function. CRC Press, Boca Raton Fla.
Muller, W. E., Blumbach, B. and Muller, I. M., 1999. Evolution of the innate and adaptive immune systems: relationships between potential immune molecules in the lowest metazoan phylum (Porifera) and those in vertebrates. Transplantation. 68 (9), 1215-1227.
Musgrave, I., 2004. Evolution of the Bacterial Flagellum, in: Young, M. and Edis, T. (Eds.), Why Intelligent Design Fails: A Scientific Critique of the Neocreationism, forthcoming from Rutgers University Press, Piscataway, N. J.
Nguyen, L., Paulsen, I. T., Tchieu, J., Hueck, C. J. and Saier, M. H., Jr., 2000. Phylogenetic analyses of the constituents of Type III protein secretion systems. J Mol Microbiol Biotechnol. 2 (2), 125-144.
Nilsson, D.-E. and Pelger, S., 1994. A pessimistic estimate of the time required for an eye to evolve. Proceedings of the Royal Society of London Series B: Biological Sciences. 256, 53-58.
Noji, H., Yasuda, R., Yoshida, M. and Kinosita, K., Jr., 1997. Direct observation of the rotation of F1-ATPase. Nature. 386 (6622), 299-302. DOI link.
Nougayrede, J. P., Fernandes, P. J. and Donnenberg, M. S., 2003. Adhesion of enteropathogenic Escherichia coli to host cells. Cell Microbiology. 5 (6), 359-372. DOI link.
Novikova, N. A., Levitsky, D. I., Metlina, A. L. and Poglazov, B. F., 2000. Interaction of bacterial flagellum filaments with skeletal muscle myosin. IUBMB Life. 50 (6), 385-390. DOI link.
Ohnishi, K., Fan, F., Schoenhals, G. J., Kihara, M. and Macnab, R. M., 1997. The FliO, FliP, FliQ, and FliR proteins of Salmonella typhimurium: putative components for flagellar assembly. J Bacteriol. 179 (19), 6092-6099.
Oplatka, A., 1998a. Are rotors at the heart of all biological motors? Biochem Biophys Res Commun. 246 (2), 301-306. DOI link.
Oplatka, A., 1998b. Do the bacterial flagellar motor and ATP synthase operate as water turbines? Biochem Biophys Res Commun. 249 (3), 573-578. DOI link.
Oster, G. and Wang, H., 2003. Rotary protein motors. Trends Cell Biol. 13 (3), 114-121. DOI link.
Pasquier, L. D. and Litman, G. W. (Eds.), 2000. Origin and Evolution of the Vertebrate Immune System (Series: Current Topics in Microbiology and Immunology). Springer, Berlin.
Peabody, C. R., Chung, Y. J., Yen, M. R., Vidal-Ingigliardi, D., Pugsley, A. P. and Saier, M. H., Jr., 2003. Type II protein secretion and its relationship to bacterial type IV pili and archaeal flagella. Microbiology. 149 (Pt 11), 3051-3072. DOI link.
Pei, Z., Burucoa, C., Grignon, B., Baqar, S., Huang, X. Z., Kopecko, D. J., Bourgeois, A. L., Fauchere, J. L. and Blaser, M. J., 1998. Mutation in the peb1A locus of Campylobacter jejuni reduces interactions with epithelial cells and intestinal colonization of mice. Infect Immun. 66 (3), 938-943.
Pellmyr, O. and Krenn, H. W., 2002. Origin of a complex key innovation in an obligate insect-plant mutualism. Proc Natl Acad Sci U S A. 99 (8), 5498-5502. DOI link.
Plano, G. V., Day, J. B. and Ferracci, F., 2001. Type III export: new uses for an old pathway. Mol Microbiol. 40 (2), 284-293. DOI link.
Poethke, H. J. and Hovestadt, T., 2002. Evolution of density- and patch-size-dependent dispersal rates. Proc R Soc Lond B Biol Sci. 269 (1491), 637-645. DOI link.
Prum, R. O. and Brush, A. H., 2002. The evolutionary origin and diversification of feathers. Q Rev Biol. 77 (3), 261-295. DOI link.
Pugsley, A. P., 1993. The complete general secretory pathway in gram-negative bacteria. Microbiol Rev. 57 (1), 50-108.
Purcell, E. M., 1977. Life at low Reynolds number. American Journal of Physics. 45, 3-11.
Purcell, E. M., 1997. The efficiency of propulsion by a rotating flagellum. Proc Natl Acad Sci U S A. 94, 11307-11311. DOI link.
Rizzotti, M., 2000. Early Evolution: From the appearance of the first cell to the first modern organisms. Birkh�user Verlag, Boston.
Rosenhouse, J., 2002. Probability, optimization theory, and evolution. Evolution. 56 (8), 1721-1722.
Sabbert, D. and Junge, W., 1997. Stepped versus continuous rotatory motors at the molecular scale. Proc Natl Acad Sci U S A. 94 (6), 2312-2317. DOI link.
Sadowsky, M. J., Tong, Z., de Souza, M. and Wackett, L. P., 1998. AtzC is a new member of the amidohydrolase protein superfamily and is homologous to other atrazine-metabolizing enzymes. J Bacteriol. 180 (1), 152-158.
Saier, M. H., Jr., 2003. Tracing pathways of transport protein evolution. Mol Microbiol. 48 (5), 1145-1156. DOI link.
Salvini-Plawen, S. V. and Mayr, E., 1977. On the evolution of photoreceptors and eyes. Evolutionary Biology. 10, 207-263.
Sandkvist, M., 2001. Biology of type II secretion. Mol Microbiol. 40 (2), 271-283. DOI link.
Sauer, F. G., Barnhart, M., Choudhury, D., Knight, S. D., Waksman, G. and Hultgren, S. J., 2000. Chaperone-assisted pilus assembly and bacterial attachment. Curr Opin Struct Biol. 10, 548-556. DOI link.
Scharfman, A., Arora, S. K., Delmotte, P., Van Brussel, E., Mazurier, J., Ramphal, R. and Roussel, P., 2001. Recognition of Lewis x derivatives present on mucins by flagellar components of Pseudomonas aeruginosa. Infect Immun. 69 (9), 5243-5248. DOI link.
Schmitt, R., 2003. Helix rotation model of the flagellar rotary motor. Biophys J. 85 (2), 843-852.
Seffernick, J. L. and Wackett, L. P., 2001. Rapid evolution of bacterial catabolic enzymes: a case study with atrazine chlorohydrolase. Biochemistry. 40 (43), 12747-12753. DOI link.
Shah, D. S., Perehinec, T., Stevens, S. M., Aizawa, S. I. and Sockett, R. E., 2000. The flagellar filament of Rhodobacter sphaeroides: pH-induced polymorphic transitions and analysis of the fliC gene. J Bacteriol. 182 (18), 5218-5224. DOI link.
Shah, D. S. and Sockett, R. E., 1995. Analysis of the motA flagellar motor gene from Rhodobacter sphaeroides, a bacterium with a unidirectional, stop-start flagellum. Mol Microbiol. 17 (5), 961-969.
Silverman, M. and Simon, M., 1974. Flagellar rotation and the mechanism of bacterial motility. Nature. 249 (452), 73-74.
Smyth, C. J., Marron, M. B., Twohig, J. M. and Smith, S. G., 1996. Fimbrial adhesins: similarities and variations in structure and biogenesis. FEMS Immunol Med Microbiol. 16 (2), 127-139. DOI link.
Spormann, A. M., 1999. Gliding motility in bacteria: insights from studies of Myxococcus xanthus. Microbiol Mol Biol Rev. 63 (3), 621-641.
Stoodley, P., Sauer, K., Davies, D. G. and Costerton, J. W., 2002. Biofilms as complex differentiated communities. Annu Rev Microbiol. 56, 187-209. DOI link.
Suerbaum, S., 1995. The complex flagella of gastric Helicobacter species. Trends Microbiol. 3 (5), 168-171. DOI link.
Sukhan, A., Kubori, T. and Gal�n, J. E., 2003. Synthesis and localization of the Salmonella SPI-1 type III secretion needle complex proteins PrgI and PrgJ. J Bacteriol. 185 (11), 3480-3483. DOI link.
Szurmant, H., Bunn, M. W., Cannistraro, V. J. and Ordal, G. W., 2003. Bacillus subtilis hydrolyzes CheY-P at the location of its action: the flagellar switch. J Biol Chem. 14, 14.
Thanassi, D. G., 2002. Ushers and secretins: channels for the secretion of folded proteins across the bacterial outer membrane. J Mol Microbiol Biotechnol. 4 (1), 11-20.
Thanassi, D. G. and Hultgren, S. J., 2000a. Multiple pathways allow protein secretion across the bacterial outer membrane. Curr Opin Cell Biol. 12 (4), 420-430. DOI link.
Thanassi, D. G. and Hultgren, S. J., 2000b. Assembly of complex organelles: pilus biogenesis in gram-negative bacteria as a model system. Methods. 20 (1), 111-126. DOI link.
Thanassi, D. G., Saulino, E. T. and Hultgren, S. J., 1998. The chaperone/usher pathway: a major terminal branch of the general secretory pathway. Curr Opin Microbiol. 1 (2), 223-231. DOI link.
Thar, R. and Kuhl, M., 2002. Conspicuous veils formed by vibrioid bacteria on sulfidic marine sediment. Appl Environ Microbiol. 68 (12), 6310-6320. DOI link.
Thomas, N. A., Bardy, S. L. and Jarrell, K. F., 2001. The archaeal flagellum: a different kind of prokaryotic motility structure. FEMS Microbiol Rev. 25 (2), 147-174. DOI link.
Thomas, N. A., Mueller, S., Klein, A. and Jarrell, K. F., 2002. Mutants in flaI and flaJ of the archaeon Methanococcus voltae are deficient in flagellum assembly. Mol Microbiol. 46 (3), 879-887. DOI link.
Thornhill, R. H. and Ussery, D. W., 2000. A classification of possible routes of Darwinian evolution. J Theor Biol. 203 (2), 111-116. DOI link.
Trachtenberg, S., Gilad, R. and Geffen, N., 2003. The bacterial linear motor of Spiroplasma melliferum BC3: from single molecules to swimming cells. Mol Microbiol. 47 (3), 671-697. DOI link.
True, J. R. and Carroll, S. B., 2002. Gene co-option in physiological and morphological evolution. Annu Rev Cell Dev Biol. 18, 53-80. DOI link.
van Wely, K. H., Swaving, J., Freudl, R. and Driessen, A. J., 2001. Translocation of proteins across the cell envelope of Gram-positive bacteria. FEMS Microbiol Rev. 25 (4), 437-454. DOI link.
Velicer, G. J., Lenski, R. E. and Kroos, L., 2002. Rescue of social motility lost during evolution of Myxococcus xanthus in an asocial environment. J Bacteriol. 184 (10), 2719-2727.
Vogel, S., 1988. Life's devices: the physical world of animals and plants. Princeton University Press, Princeton, NJ.
Vogel, S., 1994. Life in Moving Fluids. Princeton University Press, Princeton, NJ.
Vogler, A. P., Homma, M., Irikura, V. M. and Macnab, R. M., 1991. Salmonella typhimurium mutants defective in flagellar filament regrowth and sequence similarity of FliI to F0F1, vacuolar, and archaebacterial ATPase subunits. J Bacteriol. 173 (11), 3564-3572.
Walz, D. and Caplan, S. R., 2002. Bacterial flagellar motor and H(+)/ATP synthase: two proton-driven rotary molecular devices with different functions. Bioelectrochemistry. 55 (1-2), 89-92. DOI link.
Weber, J., Muharemagic, A., Wilke-Mounts, S. and Senior, A. E., 2003a. F1F0-ATP synthase. Binding of delta subunit to a 22-residue peptide mimicking the N-terminal region of alpha subunit. J Biol Chem. 278 (16), 13623-13626. DOI link.
Weber, J. and Senior, A. E., 2003. ATP synthesis driven by proton transport in F1F0-ATP synthase. FEBS Lett. 545 (1), 61-70. DOI link.
Weber, J., Wilke-Mounts, S. and Senior, A. E., 2003b. Identification of the F1-binding surface on the delta-subunit of ATP synthase. J Biol Chem. 278 (15), 13409-13416. DOI link.
Whitesides, G. M., 2001. The once and future nanomachine. Scientific American. 285 (3), 78-83.
Whitman, W. B., Coleman, D. C. and Wiebe, W. J., 1998. Prokaryotes: The unseen majority. Proc Natl Acad Sci U S A. 95, 6578-6583. DOI link.
Wilkens, S. and Capaldi, R. A., 1998. Solution structure of the epsilon subunit of the F1-ATPase from Escherichia coli and interactions of this subunit with beta subunits in the complex. J Biol Chem. 273 (41), 26645-26651. DOI link.
Wu, H. and Fives-Taylor, P. M., 2001. Molecular strategies for fimbrial expression and assembly. Crit Rev Oral Biol Med. 12 (2), 101-115.
Yamashiro, T., Nakasone, N., Honma, Y., Albert, M. J. and Iwanaga, M., 1994. Purification and characterization of Vibrio cholerae O139 fimbriae. FEMS Microbiol Lett. 115 (2-3), 247-252. DOI link.
Youderian, P., Burke, N., White, D. J. and Hartzell, P. L., 2003. Identification of genes required for adventurous gliding motility in Myxococcus xanthus with the transposable element mariner. Mol Microbiol. 49 (2), 555-570. DOI link.
Young, G. M., Schmiel, D. H. and Miller, V. L., 1999. A new pathway for the secretion of virulence factors by bacteria: the flagellar export apparatus functions as a protein-secretion system. Proc Natl Acad Sci U S A. 96 (11), 6456-6461. DOI link.
Young, K. D., 2001. Peptidoglycan, Nature Encyclopedia of Life Sciences, Nature Publishing Group, London. DOI link.
Zaar, A., Fuchs, G., Golecki, J. R. and Overmann, J., 2003. A new purple sulfur bacterium isolated from a littoral microbial mat, Thiorhodococcus drewsii sp. nov. Arch Microbiology. 179 (3), 174-183.
Zhai, Y. F., Heijne, W. and Saier, M. H., 2003. Molecular modeling of the bacterial outer membrane receptor energizer, ExbBD/TonB, based on homology with the flagellar motor, MotAB. Biochim Biophys Acta. 1614 (2), 201-210. DOI link.
Zhulin, I. B., Nikolskaya, A. N. and Galperin, M. Y., 2003. Common extracellular sensory domains in transmembrane receptors for diverse signal transduction pathways in bacteria and archaea. J Bacteriol. 185 (1), 285-294. DOI link.